|
|






 Arabic
Arabic Bengali
Bengali Chinese
Chinese English
English French
French German
German Hebrew
Hebrew Hindi
Hindi Italian
Italian Japanese
Japanese Korean
Korean Malay
Malay Polish
Polish Portuguese
Portuguese Spanish
Spanish Turkish
Turkish Ukrainian
Ukrainian Vietnamese
Vietnamese
Notatki z wykładów, ściągawki
Chemia nieorganiczna. Ściągawka: krótko, najważniejsza
Katalog / Notatki z wykładów, ściągawki Spis treści
1. Związek między procesami przemiany materii i energii w organizmie. Układ termodynamiczny Procesy życiowe na Ziemi są w dużej mierze spowodowane akumulacją energii słonecznej w substancjach biogennych (białka, tłuszcze, węglowodany) i późniejszymi przemianami tych substancji w organizmach żywych z uwolnieniem energii. Prace A.M. Lavoisiera (1743-1794) i P.S. Laplace (1749-1827) wykazały, poprzez bezpośrednie pomiary kalorymetryczne, że energia uwalniana w procesie życia determinowana jest utlenianiem produktów spożywczych przez tlen w powietrzu wdychanym przez zwierzęta. Wraz z rozwojem w XIX-XX wieku. termodynamiki, stało się możliwe ilościowe obliczanie konwersji energii w reakcjach biochemicznych i przewidywanie ich kierunku. Metoda termodynamiczna opiera się na szeregu ścisłych pojęć: „układ”, „stan układu”, „energia wewnętrzna układu”, „funkcja stanu układu”. układ termodynamiczny wywoływany jest dowolny obiekt natury, składający się z wystarczająco dużej liczby cząsteczek (jednostek strukturalnych) i oddzielony od innych obiektów natury rzeczywistą lub urojoną powierzchnią graniczną (interfejs). Obiekty przyrody, które nie są objęte systemem, nazywane są środowiskiem. Najczęstszymi cechami układów są m – masa substancji zawartej w układzie oraz E – energia wewnętrzna układu. Masę substancji układu określa całkowita masa cząsteczek, z których się składa. Energia wewnętrzna układu jest sumą energii ruchu termicznego cząsteczek i energii oddziaływania między nimi. Systemy ze względu na charakter wymiany materii i energii z otoczeniem dzielą się na trzy typy: izolowane, zamknięte i otwarte. system izolowany nazywa się układ, który nie wymienia ani materii, ani energii z medium (Δm = 0, ΔE = 0). Układ zamknięty to układ, który nie wymienia materii z otoczeniem, ale może wymieniać energię (Δm = 0, ΔE^ 0). Wymiana energii może odbywać się poprzez przekazywanie ciepła lub wykonywanie pracy. otwarty system nazywa się układ, który może wymieniać zarówno materię, jak i energię z medium (Δm ≠ 0, ΔE ≠ 0). Ważnym przykładem systemu otwartego jest żywa komórka. Systemy, w zależności od stanu skupienia substancji, z której się składają, dzielą się na jednorodne i niejednorodne. W układzie jednorodnym nie występują gwałtowne zmiany właściwości fizycznych i chemicznych podczas przechodzenia z jednego obszaru układu do drugiego. Przykładem takiego systemu jest osocze krwi, które jest roztworem różnych substancji biogennych. System heterogeniczny składa się z dwóch lub więcej jednorodnych części. Przykładem układu heterogennego jest krew pełna, czyli osocze z komórkami – erytrocytami i leukocytami. 2. Pierwsza zasada termodynamiki. Pojęcia charakteryzujące system Pierwsza zasada termodynamiki dostarcza rygorystycznych ram ilościowych do analizy energii różnych systemów. Aby go sformułować, konieczne jest wprowadzenie szeregu nowych pojęć charakteryzujących system. Jednym z najważniejszych pojęć jest stan systemu. Stan rozumiany jest jako zbiór właściwości systemu, które umożliwiają zdefiniowanie systemu z punktu widzenia termodynamiki. Jako uogólnioną charakterystykę stanu systemu stosuje się następujące pojęcia: „równowaga”, „stacjonarny”, „stan przejściowy”. Stan układu nazywany jest równowagą, jeśli wszystkie właściwości pozostają stałe przez dowolny długi okres i nie ma przepływów materii i energii w układzie. Jeśli właściwości układu są stałe w czasie, ale występują przepływy materii i energii, stan nazywamy stacjonarnym. Stany rozróżnia się ilościowo za pomocą zmiennych termodynamicznych. Zmienne termodynamiczne to wielkości charakteryzujące stan układu jako całości. Nazywa się je również parametrami termodynamicznymi układu. Najważniejszymi zmiennymi termodynamicznymi są ciśnienie p, temperatura T, objętość układu V lub całkowita masa układu m, masa substancji chemicznych (składników) mk wchodzących w skład układu lub stężenie tych substancji. Należy zaznaczyć, że na podstawie podobnych cech (temperatura, masa ciała, skład płynów biologicznych, ciśnienie krwi) lekarz określa stan pacjenta. Nazywa się przejście systemu z jednego stanu do drugiego proces. W wyniku procesu zmienia się stan układu i zmienne termodynamiczne. Jeżeli oznaczymy wartość zmiennej termodynamicznej w stanie początkowym jako Х1 i wreszcie – X2 , to zmiana tej zmiennej jest odpowiednio równa ΔX = X2 -X1 i nazywa się przyrostem zmiennej termodynamicznej X. Przyrost przyjęty z przeciwnym znakiem nazywa się spadkiem zmiennej X. Energia wewnętrzna układu E jest jedną z funkcji termodynamicznych stanu. Ważną cechą funkcji stanu jest ich niezależność od sposobu osiągnięcia danego stanu systemu. Zmiana energii wewnętrznej układu ΔE wynika z pracy W, która jest wykonywana podczas interakcji układu z otoczeniem, oraz wymiany ciepła Q między otoczeniem a układem, stosunek między tymi wielkościami wynosi treść I zasady termodynamiki. Wzrost energii wewnętrznej układu ΔE w pewnym procesie jest równy ciepłu Q otrzymanemu przez układ plus praca W wykonana na układzie w tym procesie: ∆E=Q+W. W systemach biologicznych ciepło jest zwykle oddawane przez system do środowiska zewnętrznego, a praca jest wykonywana przez system z powodu utraty energii wewnętrznej. Wygodnie jest przedstawić matematyczny zapis pierwszej zasady termodynamiki w postaci: ∆E = Q - W. Wszystkie wielkości w powyższych wzorach są mierzone w dżulach (J). 3. Pierwsza zasada termodynamiki Pierwsze prawo termodynamiki jest jednym z podstawowych praw natury, którego nie można wyprowadzić z żadnych innych praw. O jej słuszności świadczą liczne eksperymenty, w szczególności nieudane próby zbudowania perpetuum mobile pierwszego rodzaju, czyli takiej, która mogłaby pracować przez dowolnie długi czas bez dostarczania energii z zewnątrz. W zależności od warunków procesu w układzie stosuje się różne funkcje stanu, które wywodzą się z I zasady termodynamiki. Jednocześnie zamiast złożonych systemów biologicznych, do wyciągania wniosków na temat przemian masy i energii stosuje się uproszczone modele. Ciśnienie w układzie jest utrzymywane na stałym poziomie, równe ciśnieniu zewnętrznemu. Takie procesy zachodzące przy p = const nazywane są izobarycznymi. Wiadomo, że prace związane z ekspansją wykonane w procesie izobarycznym to: W = ρΔV, gdzie ΔV jest przyrostem objętości układu, równym różnicy między objętościami w stanach 2 i 1. Zastępując pracę rozwinięcia matematycznym wyrażeniem pierwszego prawa i wykonując proste przekształcenia otrzymujemy: Qρ = ΔE + pΔV = (E2 + ρV2) - (E1 +ρΔV1) gdzie Qρ jest ciepłem procesu izobarycznego; 1, 2 - indeksy związane z początkiem i końcem procesu. Wartość (E + pV) jest funkcją stanu układu, oznaczaną przez H i nazywaną entalpią: H = mi + ρV. W związku z tym wyrażenie można zapisać jako: Qp = H2 - H1 = ∆H. Z tego wyrażenia wynika, że entalpia - funkcja stanu, której przyrost jest równy ciepłu odbieranemu przez układ w procesie izobarycznym. Pomiar przyrostu entalpii w pewnym procesie można przeprowadzić przeprowadzając ten proces w kalorymetrze przy stałym ciśnieniu. W ten sposób A.M. Lavoisier i P.S. Laplace przeprowadzili swoje eksperymenty, badając energetykę metabolizmu w żywym organizmie. W przypadkach, gdy zmiana stanu układu następuje przy stałej objętości, proces nazywa się izochorycznym. W tym przypadku zmiana objętości AV jest równa zeru i zgodnie ze wzorem praca rozszerzania W = 0. Następnie z matematycznego wyrażenia pierwszej zasady termodynamiki wynika: Qv = E. Definicja termodynamiczna wynika z powyższej zależności: energia wewnętrzna - funkcja stanu, której przyrost jest równy QV ciepła uzyskanego przez układ w procesie izochorycznym. Dlatego zmianę energii wewnętrznej w pewnym procesie można zmierzyć, przeprowadzając ten proces w kalorymetrze przy stałej objętości. Wynika z tego, że przy ρ = const przyrosty energii wewnętrznej i entalpii są powiązane zależnością: ∆H = ∆E + ρ∆V. 4. Prawo Hessa Dział termodynamiki, który bada przemiany energii w reakcjach chemicznych, nazywa się termodynamiką chemiczną. Równanie reakcji, dla którego wskazane są zmiany energii wewnętrznej ΔE, entalpii ΔH lub innej funkcji stanu odpowiadającej tej reakcji, nazywa się termochemicznym. Reakcje chemiczne, podczas których entalpia układu spada (ΔH < 0) i ciepło jest uwalniane do środowiska zewnętrznego, nazywane są egzotermiczny. Reakcje, w których wzrasta entalpia (ΔH > 0) i układ pochłania ciepło Qp na zewnątrz nazywają się endotermiczny. Utlenianie glukozy tlenem następuje z uwolnieniem dużej ilości ciepła (Qp = 2800 kJ/ /mol), czyli proces ten jest egzotermiczny. Odpowiednie równanie termochemiczne zostanie zapisane jako С6 Н12 О6 + 602 = 6С02 + 6H2Och, ΔH = 2800 kJ. Reakcjom zachodzącym w roztworze zwykle towarzyszy niewielka zmiana objętości układu, tj. ΔV ≈ 0. W związku z tym w wielu przypadkach w obliczeniach biologicznych można przyjąć, że ΔH = ΔE. W konsekwencji wydzielanie ciepła w takich układach wynika głównie ze spadku energii wewnętrznej w wyniku reakcji i odwrotnie. Entalpia tworzenia związku A jest zmianą entalpii układu ΔHA towarzyszące tworzeniu 1 mola związku A z prostych substancji. Zakłada się, że entalpie tworzenia tlenu, węgla, wodoru i wszystkich innych pierwiastków (prostych) są równe zeru. Pozostałe rzeczy są równe, energia wewnętrzna i entalpia są proporcjonalne do ilości materii w układzie. Takie funkcje termodynamiczne nazywane są rozległymi. Z punktu widzenia termodynamiki reakcja postaci ogólnej nAA + pВ = nС + nieD , Δh jest przejściem układu ze stanu początkowego o entalpii H1 do stanu 2 z entalpią H2. Zmiana entalpii układu w wyniku tego przejścia, zwana entalpią tej reakcji, jest równa różnicy: Hpia = H2 - H1 = (rzeczcHc + nieDHD) - (NАНА + nieBHB). Prawo stałości sum ciepła zostało odkryte przez rosyjskiego chemika G. I. Hessa w 1840 roku. Jest on odkrywcą stosowalności pierwszej zasady termodynamiki w przemianach chemicznych i twórcą termodynamiki chemicznej. Obecnie prawo Hessa uważane jest za konsekwencję pierwszej zasady termodynamiki i jest sformułowane w następujący sposób: przyrost entalpii powstawania danych produktów z danych reagentów przy stałym ciśnieniu nie zależy od liczby i rodzaju reakcji prowadzących do powstania tych produktów. W obliczeniach termochemicznych częściej stosuje się nie samo prawo Hessa, ale jego konsekwencję, wyprowadzoną powyżej dla konkretnego przypadku utleniania glukozy w postaci równości (2). Dla reakcji przedstawionej w postaci ogólnej pАA + pвB = = nсC + nDD, konsekwencja prawa Hessa jest zapisana za pomocą równości ΔHpia = (rzeczCHC + nieDHD) - (NAHA ++nBHB ) i jest sformułowana w następujący sposób: entalpia reakcji jest równa sumie algebraicznej entalpii tworzenia stechiometrycznej ilości produktów minus suma algebraiczna entalpii tworzenia stechiometrycznej ilości reagentów. 5. Druga zasada termodynamiki. Energia swobodna Gibbsa Ciało wykonuje pracę, wydając energię wewnętrzną zmagazynowaną w postaci energii chemicznego oddziaływania atomów substancji składowych. Wyrażenie matematyczne ΔE = Q - W pierwszej zasady termodynamiki określa dokładną zależność pomiędzy wewnętrznym zużyciem energii układu ΔE, pracą W wykonaną przez układ i energią Q traconą w postaci ciepła. Jednak na podstawie pierwszej zasady termodynamiki nie można określić, jaka część pobranej energii wewnętrznej może zostać zamieniona na pracę. Teoretyczne szacunki kosztów opierają się na drugiej zasadzie termodynamiki. Prawo to nakłada surowe ograniczenia na efektywność przetwarzania energii w pracę, a ponadto pozwala wprowadzić kryteria możliwości spontanicznego przebiegu procesu. Proces nazywa się spontanicznyjeśli odbywa się to bez żadnych wpływów, gdy system jest pozostawiony sam sobie. Istnieją procesy, w których energia wewnętrzna układu się nie zmienia (ΔE = 0). Takie procesy obejmują na przykład jonizację kwasu octowego w wodzie. Wraz ze wzrostem energii wewnętrznej (ΔE > 0) zachodzi szereg spontanicznych procesów. Obejmuje to w szczególności typowe reakcje tworzenia bionieorganicznych związków albuminy (białka osocza krwi) z jonami metali, takimi jak Cu2+. Zmiana energii wewnętrznej AE dla systemów zamkniętych nie może służyć jako kryterium dla procesów spontanicznych. W konsekwencji pierwsza zasada termodynamiki, z której wywodzi się to kryterium, nie wystarcza do rozwiązania kwestii spontaniczności, a także wydajności procesów. Rozwiązanie tych pytań osiąga się za pomocą drugiej zasady termodynamiki. Aby sformułować drugą zasadę termodynamiki, konieczne jest wprowadzenie pojęć procesów odwracalnych i nieodwracalnych w sensie termodynamicznym. Jeżeli układ jest w równowadze, stan ten utrzymuje się w nieskończoność w tych samych warunkach zewnętrznych. Gdy zmieniają się warunki zewnętrzne, stan systemu może się zmienić, tzn. w systemie może zachodzić proces. Mówi się, że proces jest termodynamicznie odwracalny, jeśli podczas przejścia ze stanu początkowego 1 do stanu końcowego 2 wszystkie stany pośrednie są w równowadze. Proces nazywa się termodynamicznie nieodwracalnym, jeśli przynajmniej jeden ze stanów pośrednich jest nierównowagowy. Proces odwracalny można przeprowadzić jedynie przy dostatecznie powolnej zmianie parametrów układu - temperatury, ciśnienia, stężenia substancji itp. Szybkość zmian parametrów musi być taka, aby odchylenia od równowagi powstałe w trakcie procesu były znikome. Należy zaznaczyć, że odwracalność wiąże się z ważnym problemem medycznym – konserwacją tkanek w niskich temperaturach. Procesy odwracalne to graniczny przypadek rzeczywistych procesów zachodzących w przyrodzie i realizowanych w przemyśle lub laboratoriach. 6. Druga zasada termodynamiki. Entropia Maksymalna praca Wмакс, który można uzyskać przy danej utracie energii wewnętrznej ΔE w procesie przejścia ze stanu 1 do stanu 2, jest osiągany tylko wtedy, gdy proces ten jest odwracalny. Zgodnie z wyrażeniem na pierwszą zasadę termodynamiki ciepło minimalne Qmin Qmin \uXNUMXd ΔE - Wмакс . Maksymalny osiągalny współczynnik sprawności, charakteryzujący efektywność kosztową energii wewnętrznej układu, jest odpowiednio równy ηмакс=Wмакс / E. W nieodwracalnym procesie przejścia ze stanu 1 do stanu 2 praca wykonana przez system jest mniejsza niż W. Aby obliczyć maksymalny współczynnik hмакс przy znanej wartości ΔE konieczne jest poznanie wartości Wмакс lub Qmin Wмакс = ΔE - Qmin , zatem ηмакс = 1 - ΔE / Qmin . Wartość Qmin można obliczyć z drugiej zasady termodynamiki za pomocą funkcji stanu termodynamicznego zwanej entropią. Pojęcie entropii zostało wprowadzone (1865) przez niemieckiego fizyka R. Yu Clausiusa (1822-1888), jednego z twórców termodynamiki i molekularnej teorii kinetycznej procesów termicznych. Termodynamiczna definicja entropii według Clausiusa: entropia jest funkcją stanu, której przyrost ΔS jest równy ciepłu Qmin doprowadzone do układu w odwracalnym procesie izotermicznym, podzielone przez temperaturę bezwzględną T, w której proces jest realizowany: ∆S = Qmin / T. Ze wzoru wynika, że jednostką entropii jest J/K. Przykładem odwracalnego procesu izotermicznego jest powolne topienie lodu w termosie z wodą o temperaturze 273°K. Ustalono doświadczalnie, że do stopienia 1 mola lodu (18 g) należy dostarczyć co najmniej 6000 J ciepła. W tym przypadku entropia układu woda-lód w termosie wzrasta o ΔS = 6000 J: 273°K = 22 J/K. Gdy termos z wodą zostanie schłodzony do 273°K, 6000 J ciepła można powoli usunąć, a podczas krystalizacji wody powstaje 1 mol lodu. W tym procesie Qmin ma we wzorze wartość ujemną. W związku z tym entropia układu lód-woda po utworzeniu 1 mola lodu zmniejsza się o ΔS = 22 J/K. Podobnie można obliczyć zmianę entropii dla dowolnych izotermicznych procesów fizykochemicznych, jeśli znane jest ciepło dostarczane do układu lub od niego odprowadzane podczas tych procesów. Jak wiadomo z fizyki, ciepło to można wyznaczyć za pomocą pomiarów kalorymetrycznych. Zatem zmiana entropii, a także dwie inne funkcje stanu układu - energia wewnętrzna i entalpia, są wielkością określoną eksperymentalnie. Fizyczne znaczenie entropii, a także energii wewnętrznej, wyraźnie ujawnia się, gdy rozważamy procesy zachodzące w układach izolowanych z molekularnego punktu widzenia kinetyki. 7. Formuła Boltzmanna Systemy izolowane z definicji nie wymieniają ani materii, ani energii z otoczeniem. Oczywiście takie systemy tak naprawdę nie istnieją w naturze. Jednak bardzo dobrą izolację można uzyskać umieszczając system w termosie zamkniętym korkiem. Okazuje się, że każdy spontaniczny proces może zachodzić w układzie izolowanym tylko wtedy, gdy charakteryzuje się wzrostem entropii; w równowadze entropia układu jest stała: S ≥ 0. To stwierdzenie, oparte na obserwacjach eksperymentalnych, jest jednym z możliwych sformułowań drugiej zasady termodynamiki. Proces odwrotny do spontanicznego, zgodnie z drugą zasadą termodynamiki, nie może przebiegać w układzie izolowanym, ponieważ charakteryzuje się spadkiem entropii. Badanie różnych układów izolowanych pokazuje, że procesy spontaniczne są zawsze związane ze wzrostem liczby mikrostanów w układzie. W tych samych procesach wzrasta entropia S układu, tj. entropia wzrasta wraz ze wzrostem liczby mikrostanów. Po raz pierwszy istnienie takiej zależności zauważył austriacki fizyk L. Boltzmann, który w 1872 roku zaproponował relację: КБ =R/NA = 1,38 - 1023 J/K, gdzie KБ - stała Boltzmanna, równa stosunkowi stałej gazowej R do stałej Avogadro NA . Ta relacja nazywana jest formułą Boltzmanna. Wzór Boltzmanna pozwala teoretycznie obliczyć entropię układu z liczby jego możliwych mikrostanów. Takie obliczenia są zgodne z wartościami wyznaczonymi eksperymentalnie. W szczególności wiadomo, że liczba mikrostanów substancji krystalicznych przy 0°K jest bliska w0 „1. W ten sposób można określić wartości bezwzględne entropii substancji krystalizujących, w przeciwieństwie do energii wewnętrznej E i entalpii H, dla których można określić tylko wartości względne. Wzrost liczby mikrostanów układu w wielu przypadkach może być związany ze wzrostem nieporządku w tym układzie, z przejściem do bardziej prawdopodobnych rozkładów energii układu. Na podstawie relacji Boltzmanna można podać molekularno-kinetyczną definicję entropii. Entropia jest miarą prawdopodobieństwa, że system znajduje się w danym stanie lub miarą zaburzenia systemu. Znaczenie pojęcia entropii wynika z faktu, że na podstawie tej wartości można przewidzieć kierunek procesów spontanicznych. Jednak przydatność pomiaru entropii jako kryterium kierunku procesów ogranicza się do układów izolowanych zgodnie z sformułowaniem drugiej zasady termodynamiki. 8. Energia Gibbsa Jako kryterium spontaniczności procesów w układach otwartych i zamkniętych wprowadzono nową funkcję stanu – energię Gibbsa. Funkcja ta została nazwana na cześć wielkiego amerykańskiego fizyka D. W. Gibbsa (1839-1903), który wyprowadził tę funkcję, a następnie wykorzystał ją w pracach termodynamicznych. Energia Gibbsa jest wyznaczana w kategoriach entalpii H i entropii S z zależności: G = H - S, ∆G = AH - ∆S. Na podstawie energii Gibbsa drugą zasadę termodynamiki można sformułować w następujący sposób: w izobarycznych warunkach izotermicznych (p, T = const) tylko takie procesy mogą spontanicznie zachodzić w układzie, w wyniku czego energia Gibbsa układu maleje (ΔG < 0).W stanie równowagi energia Gibbsa układu system się nie zmienia (G = const, AG = 0). ΔG < 0, p, T = const. Z powyższego wynika, że energia Gibbsa odgrywa ważną rolę w badaniu procesów bioenergetycznych. Dzięki tej funkcji stanu można przewidzieć kierunek procesów spontanicznych w układach biologicznych i obliczyć maksymalną osiągalną wydajność. Energia Gibbsa G, podobnie jak entalpia H, jest funkcją stanu układu. Dlatego zmianę energii Gibbsa ΔG można wykorzystać do scharakteryzowania przemian chemicznych w sposób podobny do zmiany entalpii ΔH. Równania reakcji, dla których wskazana jest zmiana energii Gibbsa odpowiadająca tym reakcjom, są również nazywane termochemicznymi. Reakcje chemiczne, podczas których energia Gibbsa układu spada (ΔG < 0) i praca jest wykonywana, nazywamy egzergonicznymi. Reakcje, w wyniku których wzrasta energia Gibbsa (ΔG > 0) i wykonywana jest praca nad układem, nazywane są endergonicznymi. Wywodząca się z drugiej zasady termodynamiki, energia Gibbsa jest funkcją stanu. Dlatego, podobnie jak w przypadku entalpii, prawo Hessa dla energii Gibbsa można sformułować w następującej postaci: zmiana energii Gibbsa podczas powstawania danych produktów z danych reagentów przy stałym ciśnieniu i temperaturze nie zależy od liczby i rodzaju reakcji prowadzących do powstania tych produktów. Ważnym przykładem zastosowania prawa Hessa jest obliczenie energii Gibbsa reakcji utleniania glukozy tlenem ditlenowym. Zmiana energii Gibbsa w tej reakcji przy p = 101 kPa i T = 298°K, wyznaczona na zewnątrz ciała, wynosi ΔG° = 2880 kJ/mol. Odpowiednie równanie termochemiczne jest zapisane jako: С6Н12О6 + 62 = 6 CO2 + 6H2Och, Gpia° = 2880 kJ/mol. W komórkach organizmu reakcja ta przebiega przez szereg kolejnych etapów badanych przez biochemików. Na podstawie prawa Hessa można przewidzieć, że suma zmian energii Gibbsa we wszystkich reakcjach pośrednich wynosi ΔGpia: G1 +ΔG2 +ΔG3 + … + ∆Gn = ∆Gpia ° Energia Gibbsa reakcji jest równa algebraicznej sumie energii Gibbsa tworzenia stechiometrycznej ilości produktów minus algebraiczna suma energii Gibbsa tworzenia stechiometrycznej ilości reagentów: Gpia = (rzeczcGc + nieDGD)(NAGA + nieBGB). 9. Rozwiązania. Klasyfikacja rozwiązań W zależności od stanu skupienia roztwory mogą być gazowe, ciekłe i stałe. Każde rozwiązanie składa się z substancji rozpuszczonych i rozpuszczalnika, chociaż te koncepcje są nieco arbitralne. Na przykład, w zależności od stosunku ilości alkoholu do wody, układ ten może być roztworem alkoholu w wodzie lub wody w alkoholu. Zwykle za rozpuszczalnik uważa się składnik znajdujący się w roztworze w tym samym stanie agregacji, co przed rozpuszczeniem. Badanie roztworów budzi szczególne zainteresowanie lekarzy, ponieważ najważniejszymi płynami biologicznymi – krew, limfa, mocz, ślina, pot – są roztwory soli, białek, węglowodanów, lipidów w wodzie. Płyny biologiczne biorą udział w transporcie składników odżywczych (tłuszcze, aminokwasy, tlen), leków do narządów i tkanek, a także w wydalaniu metabolitów (mocznik, bilirubina, dwutlenek węgla itp.) z organizmu. Osocze krwi jest pożywką dla komórek - limfocytów, erytrocytów, płytek krwi. W płynnych ośrodkach organizmu utrzymuje się stałość kwasowości, stężenie soli i substancji organicznych. Ta stałość nazywa się homeostazą koncentracji. Klasyfikacja rozwiązań Roztwory substancji o masie molowej mniejszej niż 5000 g/mol nazywane są roztworami związków o niskiej masie cząsteczkowej (LMC), a roztwory substancji o masie molowej większej niż 5000 g/mol nazywane są roztworami związków o dużej masie cząsteczkowej ( HMC). W oparciu o obecność lub brak dysocjacji elektrolitycznej roztwory NMS dzieli się na dwie klasy - roztwory elektrolitów i nieelektrolitów. Roztwory elektrolitów - roztwory soli, kwasów, zasad, amfolitów dysocjujących na jony. Na przykład rozwiązania KNO3, HCl, KOH, Al(OH)3 , glicyna. Przewodność elektryczna roztworów elektrolitów jest wyższa niż rozpuszczalnika. Roztwory nieelektrolitów - roztwory substancji, które praktycznie nie dysocjują w wodzie. Na przykład roztwory sacharozy, glukozy, mocznika. Przewodność elektryczna roztworów nieelektrolitowych różni się niewiele od przewodności rozpuszczalnika. Roztwory NMS (elektrolity i nieelektrolity) nazywane są prawdziwymi, w przeciwieństwie do roztworów koloidalnych. Prawdziwe roztwory charakteryzują się jednorodnym składem i brakiem granicy faz między substancją rozpuszczoną a rozpuszczalnikiem. Wielkość rozpuszczonych cząstek (jonów i cząsteczek) jest mniejsza niż 109m Większość wkładek wewnątrzmacicznych to polimery, których cząsteczki (makrocząsteczki) składają się z dużej liczby powtarzających się grup lub jednostek monomerycznych połączonych wiązaniami chemicznymi. Roztwory wewnątrzmaciczne nazywane są roztworami polielektrolitowymi. Polielektrolity obejmują polikwasy (heparyna, kwas poliadenylowy, kwas poliasparaginowy itp.), polizasady (polilizyna), poliamfolity (białka, kwasy nukleinowe). Właściwości rozwiązań HMS różnią się znacznie od właściwości rozwiązań NMS. Dlatego zostaną omówione w osobnej sekcji. Rozdział ten poświęcony jest roztworom elektrolitów małocząsteczkowych, amfolitów i nieelektrolitów. 10. Woda jako rozpuszczalnik Najpopularniejszym rozpuszczalnikiem na naszej planecie jest woda. Ciało przeciętnego człowieka ważącego 70 kg zawiera około 40 kg wody. Jednocześnie na płyn wewnątrz komórek opada około 25 kg wody, a 15 kg to płyn pozakomórkowy, na który składają się osocze krwi, płyn międzykomórkowy, płyn mózgowo-rdzeniowy, płyn wewnątrzgałkowy oraz płynna treść przewodu pokarmowego. W organizmach zwierzęcych i roślinnych zawartość wody zwykle przekracza 50%, aw niektórych przypadkach zawartość wody sięga 90-95%. Ze względu na swoje anomalne właściwości woda jest wyjątkowym rozpuszczalnikiem, doskonale przystosowanym do życia. Przede wszystkim woda dobrze rozpuszcza związki jonowe i wiele związków polarnych. Ta właściwość wody wynika w dużej mierze z jej wysokiej stałej dielektrycznej (78,5). Inną dużą klasą substancji, które są dobrze rozpuszczalne w wodzie, są takie polarne związki organiczne, jak cukry, aldehydy, ketony i alkohole. Ich rozpuszczalność w wodzie tłumaczy się tendencją cząsteczek wody do tworzenia wiązań polarnych z polarnymi grupami funkcyjnymi tych substancji, na przykład z grupami hydroksylowymi alkoholi i cukrów lub z atomem tlenu grupy karbonylowej aldehydów i ketonów. Poniżej przedstawiono przykłady wiązań wodorowych ważnych dla rozpuszczalności substancji w układach biologicznych. Woda ze względu na swoją wysoką polarność powoduje hydrolizę substancji. Ponieważ woda jest główną częścią wewnętrznego środowiska organizmu, zapewnia procesy wchłaniania, przemieszczania składników odżywczych i produktów przemiany materii w organizmie. Należy zaznaczyć, że woda jest końcowym produktem biologicznego utleniania substancji, w szczególności glukozy. Powstawaniu wody w wyniku tych procesów towarzyszy wyzwolenie dużej ilości energii – około 29 kJ/mol. Ważne są również inne anomalne właściwości wody: wysokie napięcie powierzchniowe, niska lepkość, wysoka temperatura topnienia i wrzenia oraz wyższa gęstość w stanie ciekłym niż w stanie stałym. Woda charakteryzuje się obecnością asocjatów - grup cząsteczek połączonych wiązaniami wodorowymi. W zależności od powinowactwa do wody, grupy funkcyjne rozpuszczonych cząstek dzielą się na hydrofilowe (przyciągające wodę), łatwo solwatowane przez wodę, hydrofobowe (odpychające wodę) i amfifilowe. Do grup hydrofilowych zalicza się polarne grupy funkcyjne: hydroksyl -OH, amino -NH2 , tiol -SH, karboksyl -COOH. Do hydrofobowych - niepolarnych grup, np. rodniki węglowodorowe: CHXNUMX-(CH2)п -, Z6Н5 -. Substancje hifilowe obejmują substancje (aminokwasy, białka), których cząsteczki zawierają obie grupy hydrofilowe (-OH, -NH2 , -SH, -COOH) i grupy hydrofobowe: (CH3 - (CZ2)п ,-Z6Н5-). Po rozpuszczeniu substancji amfifilowych struktura wody zmienia się w wyniku oddziaływania z grupami hydrofobowymi. Zwiększa się stopień uporządkowania cząsteczek wody znajdujących się w pobliżu grup hydrofobowych, a kontakt cząsteczek wody z grupami hydrofobowymi jest minimalizowany. Grupy hydrofobowe, kojarzące się, wypychają cząsteczki wody poza obszar ich lokalizacji. 11. Stężenie roztworu i jak to wyrazić Rozwiązanie Jednorodny układ o zmiennym składzie dwóch lub więcej substancji w stanie równowagi to tzw. Substancje tworzące roztwór nazywane są składnikami roztworu. Ważną cechą roztworu jest jego stężenie. Ta wartość określa wiele właściwości rozwiązania. Stężenie substancji (składnik roztworu) to ilość mierzona ilością substancji rozpuszczonej zawartej w określonej masie lub objętości roztworu lub rozpuszczalnika. Najczęściej stosowanymi sposobami wyrażania stężenia są: ułamek masowy, stężenie molowe, stężenie równoważnika molowego, ułamek molowy, ułamek objętościowy, miano. Udział masowy W(X) wyrażone we ułamkach jednostki, procentach (%), ppm (tysięcznych procenta) oraz w częściach na milion (ppm). Ułamek masowy oblicza się według wzorów: W(X) = m(X)/m(pp), W(X) = m(X)/m(pp) × 100%, gdzie m(X) - masa danego składnika X (substancja rozpuszczona), kg (g); m (pp) to masa roztworu, kg (g). Stężenie molowe wyraża się w mol/m3 , mol/dm3 , mol/cm3 , mol/l, mol/ml. W medycynie preferowane jest stosowanie jednostek mol / l. Stężenie molowe oblicza się według wzoru: C(X) =n(X)/V(pp) = m(X)/M(X) ×V(rr), gdzie n(X) - ilość rozpuszczonej substancji w układzie, mol; M(X) jest masą molową substancji rozpuszczonej, kg/mol lub g/mol; m(X) to masa rozpuszczonej substancji, odpowiednio, kg lub g; V(rr) - objętość roztworu, l. stężenie molowe b(X) wyrażone w jednostkach mol/kg. Formularz zapisu, na przykład: b (HCl) \u0,1d XNUMX mol / kg. Oblicz stężenie molowe według wzoru: b(X) =n(X)/m(rl) = m(X)/M(X) ×m(rl) gdzie m(rl) - masa rozpuszczalnika, kg. W chemii szeroko stosowane jest pojęcie współczynnika równoważności i współczynnika równoważności. Równowartość nazywana jest cząstką rzeczywistą lub warunkową substancji X, która w danej reakcji kwasowo-zasadowej jest równoważna jednemu jonowi wodorowemu, w danej reakcji redoks - jednemu elektronowi, lub w danej reakcji wymiany pomiędzy solami - jednostce ładunku . Ułamek objętości f(X) wyrażona w ułamkach jednostkowych lub w procentach, obliczana jest według wzoru: Ф(X) = V(X)/V(rr) gdzie v(X) - objętość tego składnika X roztworu; V(rr) to całkowita objętość rozpuszczalnika. Miano roztworu jest oznaczone przez T(X), jednostka miary - kg/cm3 , g/cm3 , g/ml. Miano roztworu można obliczyć za pomocą wzoru: Т(X) = m(X)/V(rr) gdzie m(X) masa substancji, zwykle g; V(rr) objętość roztworu, ml. 12. Proces rozpuszczania Charakter procesu rozpuszczania jest złożony. Oczywiście pojawia się pytanie, dlaczego niektóre substancje są łatwo rozpuszczalne w niektórych rozpuszczalnikach, a słabo lub praktycznie nierozpuszczalne w innych. Tworzenie rozwiązań zawsze wiąże się z pewnymi procesami fizycznymi. Jednym z takich procesów jest dyfuzja substancji rozpuszczonej i rozpuszczalnika. W wyniku dyfuzji cząsteczki (cząsteczki, jony) są usuwane z powierzchni rozpuszczonej substancji i są równomiernie rozprowadzane w całej objętości rozpuszczalnika. Dlatego przy braku mieszania szybkość rozpuszczania zależy od szybkości dyfuzji. Nie można jednak wyjaśnić nierównej rozpuszczalności substancji w różnych rozpuszczalnikach wyłącznie procesami fizycznymi. Wielki rosyjski chemik D. I. Mendelejew (1834-1907) uważał, że procesy chemiczne odgrywają ważną rolę w rozpuszczaniu. Udowodnił istnienie wodzianów kwasu siarkowego H2SO4H2OH2SO42H2OH2SO44H2O i kilka innych substancji, na przykład C2Н5OH3H2A. W takich przypadkach rozpuszczaniu towarzyszy tworzenie wiązań chemicznych pomiędzy cząsteczkami substancji rozpuszczonej i rozpuszczalnika. Proces ten nazywa się solwatacją, w szczególnym przypadku, gdy rozpuszczalnikiem jest woda – hydratacją. Jak ustalono, w zależności od charakteru rozpuszczonej substancji, solwaty (hydraty) mogą powstawać w wyniku oddziaływań fizycznych: oddziaływania jon-dipol (na przykład podczas rozpuszczania substancji o strukturze jonowej (NaCl itp.). ); oddziaływanie dipol-dipol - podczas rozpuszczania substancji o strukturze molekularnej (substancje organiczne )). Oddziaływania chemiczne są przeprowadzane dzięki wiązaniom donor-akceptor. Tutaj jony substancji rozpuszczonej są akceptorami elektronów, a rozpuszczalniki (Н2Och, NH3) - donory elektronów (na przykład tworzenie kompleksów wodnych), a także w wyniku tworzenia wiązań wodorowych (na przykład rozpuszczanie alkoholu w wodzie). Dowodem na chemiczne oddziaływanie substancji rozpuszczonej z rozpuszczalnikiem są efekty termiczne i zmiana koloru towarzysząca rozpuszczaniu. Na przykład, gdy wodorotlenek potasu rozpuszcza się w wodzie, uwalniane jest ciepło: KOH + xN2O \uXNUMXd KOH (N2Oh; ΔH°rozwiązanie = 55 kJ/mol. A po rozpuszczeniu chlorku sodu ciepło jest pochłaniane: NaCl + xH2O = NaCl(H2Oh; ΔH°rozwiązanie = +3,8 kJ/mol. Ciepło uwalniane lub pochłaniane podczas rozpuszczania 1 mola substancji nazywa się ciepło roztworu Qrozwiązanie Zgodnie z pierwszą zasadą termodynamiki Qrozwiązanie = ΔHrozwiązanie, gdzie ΔHrozwiązanie jest zmianą entalpii po rozpuszczeniu danej ilości substancji. Rozpuszczenie bezwodnego białego siarczanu miedzi w wodzie prowadzi do pojawienia się intensywnego niebieskiego koloru. Powstawanie solwatów, zmiana koloru, efekty termiczne, a także szereg innych czynników, wskazują na zmianę charakteru chemicznego składników roztworu podczas jego tworzenia. Tak więc, zgodnie z nowoczesnymi koncepcjami, rozpuszczanie jest procesem fizykochemicznym, w którym rolę odgrywają zarówno fizyczne, jak i chemiczne rodzaje interakcji. 13. Termodynamika procesu rozpuszczania Zgodnie z drugą zasadą termodynamiki, przy p, T = const, substancje mogą spontanicznie rozpuszczać się w dowolnym rozpuszczalniku, jeśli w wyniku tego procesu energia Gibbsa układu zmniejszy się, tj. ΔG = (ΔН - TΔS) < 0. Wartość ΔН nazywana jest współczynnikiem entalpii, a wartość TΔS — współczynnikiem entropii rozpuszczania. Gdy substancje ciekłe i stałe są rozpuszczone, entropia układu zwykle wzrasta (ΔS > 0), ponieważ rozpuszczone substancje przechodzą z bardziej uporządkowanego stanu do mniej uporządkowanego. Udział czynnika entropii, który przyczynia się do wzrostu rozpuszczalności, jest szczególnie zauważalny w podwyższonych temperaturach, ponieważ w tym przypadku czynnik T jest duży i wartość bezwzględna iloczynu TΔS jest również duża, odpowiednio spadek wartości Gibbsa energia wzrasta. Kiedy gazy są rozpuszczone w cieczy, entropia układu zwykle spada (ΔS < 0), ponieważ substancja rozpuszczona przechodzi ze stanu mniej uporządkowanego (duża objętość) do stanu bardziej uporządkowanego (mała objętość). Spadek temperatury sprzyja rozpuszczaniu gazów, ponieważ w tym przypadku współczynnik T jest mały i wartość bezwzględna iloczynu TΔS będzie tym mniejsza, a spadek energii Gibbsa będzie większy, im mniejsza będzie wartość T. Podczas tworzenia roztworu entalpia układu może również zarówno wzrastać (NaCl), jak i spadać (KOH). Zmianę entalpii procesu rozpuszczania należy rozpatrywać zgodnie z prawem Hessa jako sumę algebraiczną wkładów endo i egzotermicznych wszystkich procesów towarzyszących procesowi rozpuszczania. Są to endotermiczne efekty zniszczenia sieci krystalicznej substancji, zerwanie wiązań cząsteczek, zniszczenie początkowej struktury rozpuszczalnika oraz egzotermiczne efekty powstawania różnych produktów interakcji, w tym solwatów. Dla uproszczenia prezentacji przyrost entalpii rozpuszczania ΔНrozwiązanie można przedstawić jako różnicę energii Ecr, wydatkowana na zniszczenie sieci krystalicznej rozpuszczonej substancji, oraz energię Esol, uwalniany podczas solwatacji cząstek substancji rozpuszczonej przez cząsteczki rozpuszczalnika. Innymi słowy, zmiana entalpii jest sumą algebraiczną zmiany entalpii ΔHcr w wyniku zniszczenia sieci krystalicznej i zmiany entalpii ΔНsol z powodu solwatacji przez cząsteczki rozpuszczalnika: ΔNrozwiązanie = ΔHcr +sol, gdzie ΔHrozwiązanie - zmiana entalpii podczas rozpuszczania. Jednak rozpuszczaniu gazów szlachetnych w rozpuszczalnikach organicznych często towarzyszy absorpcja ciepła, np. helu i neonu w acetonie, benzenie, etanolu i cykloheksanie. Podczas rozpuszczania ciał stałych o strukturze kryształu molekularnego i cieczy wiązania molekularne nie są bardzo silne, a zatem zwykle ΔHsol > ΔNcr Prowadzi to do tego, że rozpuszczanie np. alkoholi i cukrów jest procesem egzotermicznym (ΔHrozwiązanie < 0). Podczas rozpuszczania ciał stałych za pomocą sieci jonowej, stosunek energii Ecr i Esol może być inny. Jednak w większości przypadków energia uwalniana podczas solwatacji jonów nie kompensuje energii zużytej na zniszczenie sieci krystalicznej, dlatego proces rozpuszczania jest endotermiczny. W ten sposób dane termodynamiczne umożliwiają przewidywanie spontanicznego rozpuszczania różnych substancji na podstawie pierwszej i drugiej zasady termodynamiki. 14. Rozpuszczalność Jeżeli substancja rozpuszczona jest w kontakcie z rozpuszczalnikiem, proces tworzenia roztworu w wielu przypadkach przebiega samoistnie aż do osiągnięcia pewnego stężenia granicznego (występuje nasycenie). Dzieje się tak, gdy równowaga zostaje osiągnięta, gdy współczynniki entalpii i entropii są sobie równe, tj. ΔН = TΔS. Na przykład, gdy kryształy są wprowadzane do cieczy, cząsteczki lub jony przechodzą z powierzchni kryształu do roztworu. Dzięki dyfuzji cząstki są równomiernie rozłożone w całej objętości rozpuszczalnika. Rozpuszczanie przechodzi do nasycenia. Roztwór, który zawiera maksymalną ilość substancji rozpuszczonej w danej temperaturze i jest w równowadze z nadmiarem substancji rozpuszczonej, nazywa się nasyconym. Roztwór przesycony to roztwór, którego stężenie jest wyższe niż roztworu nasyconego. Roztwór o niższym stężeniu niż roztwór nasycony nazywa się nienasyconym. Zdolność substancji do rozpuszczania się w określonym rozpuszczalniku nazywa się rozpuszczalnością. Liczbowo rozpuszczalność substancji jest równa stężeniu jej nasyconego roztworu. Rozpuszczalność można wyrazić w tych samych jednostkach co stężenie, np. jako ilość substancji rozpuszczonej zawartej w 1 litrze roztworu nasyconego, mol/l, lub jako masę substancji rozpuszczonej w 100 g roztworu nasyconego . Jednostką rozpuszczalności są gramy na 100 g rozpuszczalnika. Odpowiednia wartość nazywana jest współczynnikiem rozpuszczalności. Rozpuszczalność zależy od rodzaju substancji rozpuszczonej i rozpuszczalnika, temperatury, ciśnienia i obecności innych substancji w roztworze. Wpływ na rozpuszczalność charakteru składników Zdolność substancji do rozpuszczania zależy od charakteru sił interakcji między cząsteczkami składników roztworu X1 ich2 : rozpuszczalnik - rozpuszczalnik (X1 -X1 ), substancja rozpuszczona - substancja rozpuszczona (X2 - X2 ), rozpuszczalnik - substancja rozpuszczona (X1 - X2 ) (punkty wskazują wiązanie molekularne). Rozpuszczalność substancji jest bardzo zróżnicowana. Przykłady pokazują rozpuszczalność różnych soli w tym samym rozpuszczalniku (woda) oraz rozpuszczalność tej samej substancji (AgNO3 ) w różnych rozpuszczalnikach. Substancje z wiązaniem jonowym oraz substancje składające się z cząsteczek polarnych lepiej rozpuszczają się w rozpuszczalnikach polarnych, takich jak woda, alkohole. Rozpuszczalniki te charakteryzują się wysoką stałą dielektryczną. Wysoka rozpuszczalność substancji jest często spowodowana tworzeniem się wiązań międzycząsteczkowych, w szczególności wodorowych. Zatem nieograniczoną wzajemną rozpuszczalność wody i alkoholu tłumaczy się tworzeniem wiązań wodorowych między cząsteczkami wody i alkoholu, a rozpuszczanie kryształów AgcI w wodnym roztworze amoniaku tłumaczy się tworzeniem chemicznego wiązania donor-akceptor srebra jon z cząsteczkami amoniaku (AgCI jest praktycznie nierozpuszczalny w wodzie). Z tego samego powodu pirydyna, rozpuszczalnik o niskiej stałej dielektrycznej, wykazuje bardzo wysoką rozpuszczalność. Ponieważ rozpuszczalność charakteryzuje prawdziwą równowagę, wpływ warunków zewnętrznych na ten stan (ciśnienie, temperatura) można jakościowo oszacować za pomocą zasady Le Chateliera. Takie oceny są niezbędne w praktyce nurkowania głębokiego, podczas pracy w gorących sklepach itp. 15. Rozpuszczalność gazów w cieczach. Prawa Henry-Dalton i Sechenov Rozpuszczaniu gazów w cieczach prawie zawsze towarzyszy wydzielanie ciepła. Dlatego rozpuszczalność gazów maleje wraz ze wzrostem temperatury zgodnie z zasadą Le Chateliera. Ten wzór jest często używany do usuwania rozpuszczonych gazów z wody (np. CO02) przez gotowanie. Czasami rozpuszczaniu gazu towarzyszy absorpcja ciepła (na przykład rozpuszczanie gazów szlachetnych w niektórych rozpuszczalnikach organicznych). W takim przypadku zwiększenie temperatury zwiększa rozpuszczalność gazu. Gaz nie rozpuszcza się w cieczy w nieskończoność. Przy pewnym stężeniu gazu X ustala się równowaga:  Gdy gaz rozpuszcza się w cieczy, następuje znaczny spadek objętości układu. Dlatego wzrost ciśnienia, zgodnie z zasadą Le Chateliera, powinien prowadzić do przesunięcia równowagi w prawo, czyli do wzrostu rozpuszczalności gazu. Jeżeli gaz jest słabo rozpuszczalny w danej cieczy, a ciśnienie jest niskie, to rozpuszczalność gazu jest proporcjonalna do jego ciśnienia. Zależność tę wyraża prawo Henryka (1803): ilość gazu rozpuszczonego w danej temperaturze w określonej objętości cieczy w równowadze jest wprost proporcjonalna do ciśnienia gazu. Prawo Henryka można zapisać w następującej formie: с (X) =Kr(X) ×str(X) gdzie jest stężenie gazu w roztworze nasyconym, mol/l; P(X) - ciśnienie gazu X nad roztworem, Pa; Kr(X) - stała Henry'ego dla gazu X, mol × l1 × Pa1 . Stała Henry'ego zależy od rodzaju gazu, rozpuszczalnika i temperatury. Prawo Henry'ego obowiązuje tylko dla stosunkowo rozcieńczonych roztworów, przy niskim ciśnieniu i przy braku interakcji chemicznych między cząsteczkami rozpuszczonego gazu i rozpuszczalnika. Prawo Henry'ego jest szczególnym przypadkiem ogólnego prawa Daltona. Jeśli mówimy o rozpuszczeniu nie jednej substancji gazowej, ale mieszaniny gazów, to rozpuszczalność każdego składnika jest zgodna z prawem Daltona: rozpuszczalność każdego ze składników mieszaniny gazowej w stałej temperaturze jest proporcjonalna do ciśnienia cząstkowego składnika nad cieczą i nie zależy od całkowitego ciśnienia mieszaniny i indywidualności innych składników. Innymi słowy, w przypadku rozpuszczenia mieszaniny gazów w cieczy ciśnienie cząstkowe p! ten składnik. Przez ciśnienie cząstkowe składnika rozumie się stosunek ciśnienia składnika do całkowitego ciśnienia mieszaniny gazów: Рi/ Rłącznie Ciśnienie cząstkowe składnika oblicza się według wzoru Badając rozpuszczalność gazów w cieczach w obecności elektrolitów, rosyjski fizjolog I. M. Sechenov (1829-1905) ustalił następujący wzór (prawo Sechenova): zmniejsza się rozpuszczalność gazów w cieczach w obecności elektrolitów; gazy są uwalniane. Рi = Płącznie ×(Xi) gdzie pii - ciśnienie cząstkowe składnika Xi; Рłącznie całkowite ciśnienie mieszaniny gazów; x(Xi) jest ułamkiem molowym i-tego składnika. Badając rozpuszczalność gazów w cieczach w obecności elektrolitów, rosyjski fizjolog I. M. Sechenov (1829-1905) ustalił następujący wzór (prawo Sechenova): zmniejsza się rozpuszczalność gazów w cieczach w obecności elektrolitów; gazy są uwalniane. 16. Rola dyfuzji w procesach przenoszenia substancji w układach biologicznych Dyfuzja odgrywa ważną rolę w układach biologicznych. Przede wszystkim ruch składników odżywczych i produktów przemiany materii w płynach tkankowych odbywa się poprzez dyfuzję. Ponadto w wielu przypadkach szybkość procesów fizykochemicznych w organizmach żywych jest determinowana szybkością dyfuzji reagentów, gdyż dyfuzja reagentów jest zwykle najwolniejszym etapem procesu, a reakcje biochemiczne z udziałem enzymów przebiegają bardzo szybko. Każda żywa komórka otoczona jest błoną, która służy do ochrony i regulacji środowiska wewnątrzkomórkowego. Substancje przechodzą przez błony dwoma głównymi mechanizmami: zwykłą dyfuzją (transport pasywny) oraz transferem aktywowanym energetycznie (transport aktywny). Wewnętrzna warstwa membrany składa się z łańcuchów węglowodorowych. Dlatego wiele małych obojętnych cząsteczek i niepolarnych cząsteczek HMS jest rozpuszczalnych w tej warstwie i może przechodzić przez błonę przez normalną dyfuzję wzdłuż gradientu stężenia. Taki transport substancji nazywamy pasywnym. Dyfuzja odgrywa dużą rolę w procesie dotleniania krwi w płucach. Ze względu na duże rozgałęzienia powierzchnia pęcherzyków płucnych jest duża (~ 80 m2), więc tlen aktywnie rozpuszcza się w osoczu i przedostaje się do czerwonych krwinek. Krew żylna jest pozbawiona tlenu – stężenie tlenu we krwi żylnej dąży do zera. W rezultacie gradient stężenia tlenu pomiędzy atmosferą a krwią dopływającą do płuc jest wysoki, co prowadzi do aktywnej absorpcji (zgodnie z prawem Ficka). Przenoszenie substancji z obszaru o niższym stężeniu do obszaru o wyższym stężeniu wbrew gradientowi nazywa się transportem aktywnym. Proces taki nie może zachodzić samoistnie i wymaga nakładu energii. Źródłem energii jest egzoenergetyczna reakcja hydrolizy związku bionieorganicznego – trifosforanu adenozyny (ATP). Stabilny stacjonarny rozkład stężeń jonów K wewnątrz i na zewnątrz komórki uzyskuje się, gdy przepływ jonów K przez błonę do komórki staje się równy przepływowi jonów K z komórki na skutek dyfuzji biernej. Dystrybucja (homeostaza jonów) jest podobnie osiągana dla jonów Na, tylko transport aktywny i kompensacyjna dyfuzja pasywna jonów są skierowane przeciwnie do odpowiednich przepływów jonów K. Proces dyfuzji jest szeroko stosowany w medycynie. Na przykład metoda dializy oparta na selektywności dyfuzji substancji niskocząsteczkowych przez półprzepuszczalną membranę wzdłuż gradientu stężeń jest wykorzystywana w praktyce klinicznej do stworzenia urządzenia „sztuczna nerka”. Cząsteczki wkładki domacicznej nie przechodzą przez półprzepuszczalną błonę, dlatego płyny biologiczne (na przykład osocze krwi) można oczyścić metodą dializy ze szkodliwych substancji niskocząsteczkowych - „żużli” (mocznik, kwas moczowy, bilirubina, aminy, nadmiar jonów K ), które kumulują się w różnych chorobach. Podczas oczyszczania krew pacjenta, odprowadzona z żyły, trafia do specjalnych komór z półprzepuszczalnymi membranami, przez które NMS może dyfundować i zostać usunięty z osocza. W wielu chorobach zapalnych dochodzi do niszczenia białek, aw osoczu krwi wraz z NMS znajdują się fragmenty białek (peptydy i polipeptydy), które również muszą zostać usunięte. 17. Obniżenie temperatury zamarzania i podniesienie temperatury wrzenia roztworów Bezpośrednią konsekwencją obniżenia prężności pary nad roztworem jest zmiana temperatury krzepnięcia ΔTз i temperatura wrzenia roztworów ΔТк w porównaniu z wartościami tych ilości dla czystego rozpuszczalnika. Zależności między tymi wielkościami wynikają również z drugiej zasady termodynamiki. Temperatura wrzenia cieczy to temperatura, w której prężność pary staje się równa ciśnieniu zewnętrznemu (na przykład przy 101,3 kPa temperatura wrzenia wody wynosi 100 ° C). Temperatura zamarzania (krystalizacja) cieczy to temperatura, w której prężność pary nad cieczą jest równa prężności pary nad fazą stałą. Jeśli wyznaczymy temperatury zamarzania i wrzenia roztworu T3 i Tk oraz te same wartości dla rozpuszczalnika T°3 i T°к , wtedy otrzymujemy: Тк = Тк - Temperaturaк > 0, T3 = T°3 - T3 > 0. Skutki podwyższenia temperatury wrzenia i obniżenia temperatury krzepnięcia roztworów można jakościowo wyjaśnić za pomocą zasady Le Chateliera. Rzeczywiście, jeśli w układzie równowagi „ciecz - para” (na przykład H2О(sol) - H2О(G)), wprowadzić rozpuszczalną nielotną substancję, wówczas ciśnienie pary rozpuszczalnika nad roztworem zmniejszy się. Aby zrekompensować spadek prężności pary i osiągnąć poprzedni stan równowagi, roztwór musi zostać podgrzany do wyższej temperatury (ponad 373°K), ponieważ proces jest endotermiczny. Niech będzie układ równowagi "faza stała - ciecz", na przykład H2О(t) > H2О(sol), w temperaturze 273°K. Jeśli w fazie ciekłej rozpuści się pewna ilość substancji nielotnej (nierozpuszczalnej w fazie stałej), wówczas stężenie cząsteczek wody w fazie ciekłej będzie się zmniejszać. Zgodnie z zasadą Le Chateliera rozpocznie się proces zwiększający ilość wody w fazie ciekłej – topnienie lodu. Aby ustalić nową równowagę, roztwór należy schłodzić, tj. Należy obniżyć temperaturę, ponieważ proces jest egzotermiczny. Zgodnie z prawem Raoulta dla roztworów rozcieńczonych spadek prężności pary jest proporcjonalny do stężenia roztworu. Dlatego wzrost temperatury wrzenia i spadek temperatury zamarzania takich roztworów powinien wzrastać wraz ze wzrostem ich stężenia. Badając zamrażanie i wrzenie roztworów, Raul odkrył: wzrost temperatury wrzenia ΔTк i obniżenie temperatury zamarzania ΔT3 rozcieńczone roztwory nieelektrolitów są wprost proporcjonalne do stężenia molowego roztworu: Tк = Kэb(x), T3 = Kз b(x), gdzie b(X) - stężenie molowe, mol/kg; Кз i K.э - współczynniki proporcjonalności, kg × K × mol1 , które są nazywane odpowiednio stałymi ebulliometrycznymi i kriometrycznymi. Fizyczne znaczenie stałych Kэ i K.з staje się jasne, jeśli zaakceptujemy b(X) = 1. Wtedy Kэ = ΔTк i Kз = ΔTз . Innymi słowy, stała ebuliometryczna jest liczbowo równa wzrostowi temperatury wrzenia jednomolowego roztworu, a stała kriometryczna jest równa spadkowi temperatury zamarzania jednomolowego roztworu. Stałe ebuliometryczne i kriometryczne zależą tylko od rodzaju rozpuszczalnika i nie zależą od rodzaju substancji rozpuszczonej (roztwory idealne). 18. Ciśnienie osmotyczne Osmoza to głównie jednostronne przenikanie cząsteczek rozpuszczalnika (dyfuzja) przez półprzepuszczalną membranę z rozpuszczalnika do roztworu lub z roztworu o niższym stężeniu do roztworu o wyższym stężeniu. Warunkiem koniecznym wystąpienia osmozy jest obecność rozpuszczalnika i roztworu lub dwóch roztworów o różnych stężeniach, oddzielonych półprzepuszczalną membraną. Z punktu widzenia termodynamiki siłą napędową osmozy jest chęć wyrównania stężeń układu, ponieważ w tym przypadku entropia układu wzrasta, ponieważ układ przechodzi w stan mniej uporządkowany, energia Gibbsa układu odpowiednio maleje, a potencjały chemiczne wyrównują się. Dlatego osmoza jest procesem spontanicznym. Prosty eksperyment może posłużyć za ilustrację wyjaśniającą związek między mechanizmem osmozy a zmianą prężności pary nad roztworem. Jeżeli w zamkniętym naczyniu szklanym zostanie umieszczona szklanka z czystym rozpuszczalnikiem oraz szklanka z roztworem jakiejś substancji nielotnej (poziomy płynów w naczyniach są takie same), to po chwili poziom cieczy w naczyniu jest taki sam. pierwsza szklanka zmniejszy się, a poziom roztworu w drugiej szklance wzrośnie. W tym przypadku rozpuszczalnik przechodzi z pierwszej zlewki do drugiej zlewki, co jest spowodowane (zgodnie z prawem Raoulta) niższą prężnością par rozpuszczalnika nad roztworem niż nad czystym rozpuszczalnikiem. W ten sposób przestrzeń powietrzna pomiędzy rozpuszczalnikiem a roztworem działa jak półprzepuszczalna membrana. Napełnijmy naczynie o ściankach półprzepuszczalnych wodnym roztworem glukozy i umieśćmy je w innym naczyniu z wodą tak, aby poziomy płynów w tych naczyniach się pokrywały. W wyniku osmozy zwiększa się objętość roztworu w pierwszym naczyniu i stopniowo podnosi się poziom cieczy w tym naczyniu. Tworzy to dodatkowe ciśnienie hydrostatyczne, które zapobiega osmozie. Ciśnienie hydrostatyczne kolumny cieczy w równowadze osmotycznej określa ciśnienie osmotyczne roztworu. Ciśnienie osmotyczne nazywana wartością zmierzoną przez minimalne ciśnienie hydrauliczne, które należy przyłożyć do roztworu, aby zatrzymać osmozę. Prawa ciśnienia osmotycznego. Van't Hoff zaproponował empiryczne równanie do obliczania ciśnienia osmotycznego rozcieńczonych roztworów nieelektrolitów: π = C(X)rt, gdzie π - ciśnienie osmotyczne, kPa; С(X) - stężenie molowe, mol/l; R jest uniwersalną stałą gazową równą 8,31 kPa - l / (mol - K); T to temperatura bezwzględna, K. Chociaż prawo van't Hoffa zostało ustalone na podstawie danych doświadczalnych, można je wyprowadzić z warunków równowagi termodynamicznej przy ΔG = 0. Dlatego prawo to należy traktować jako konsekwencję drugiej zasady termodynamiki. Wyrażenie w powyższej postaci jest podobne do równania Clapeyrona-Mendeleeva dla gazów doskonałych, jednak równania te opisują różne procesy. 19. Rola osmozy i ciśnienia osmotycznego w układach biologicznych Zjawisko osmozy odgrywa ważną rolę w wielu układach chemicznych i biologicznych. Osmoza reguluje przepływ wody do komórek i struktur międzykomórkowych. Elastyczność komórek (turgor), która zapewnia elastyczność tkanek i zachowanie określonego kształtu narządów, wynika z ciśnienia osmotycznego. Komórki zwierzęce i roślinne mają otoczki lub warstwę powierzchniową protoplazmy, która ma właściwości błon półprzepuszczalnych. Gdy komórki te zostaną umieszczone w roztworach o różnych stężeniach, obserwuje się osmozę. Roztwory o tym samym ciśnieniu osmotycznym nazywane są izotonicznymi. Jeżeli dwa roztwory mają różne ciśnienia osmotyczne, to roztwór o wyższym ciśnieniu osmotycznym jest hipertoniczny w stosunku do drugiego, a drugi jest hipotoniczny w stosunku do pierwszego. Komórki umieszczone w roztworze izotonicznym zachowują swój rozmiar i normalne funkcjonowanie. Po umieszczeniu komórek w roztworze hipotonicznym woda z mniej stężonego roztworu zewnętrznego przedostaje się do komórek, co prowadzi do ich pęcznienia, a następnie do pęknięcia błon i wypływu zawartości komórkowej. To zniszczenie komórek nazywa się lizą, w przypadku czerwonych krwinek proces ten nazywa się hemolizą. Krew z zawartością komórkową wydobywającą się podczas hemolizy nazywana jest krwią lakieru ze względu na swój kolor. Po umieszczeniu komórek w roztworze hipertonicznym woda opuszcza komórki w bardziej stężonym roztworze i obserwuje się marszczenie (wysychanie) komórek. Zjawisko to nazywa się plazmolizą. Ludzkie płyny biologiczne (krew, limfa, płyny tkankowe) to wodne roztwory związków o niskiej masie cząsteczkowej – NaCl, KCl, CaCl, związków o dużej masie cząsteczkowej – białek, polisacharydów, kwasów nukleinowych oraz pierwiastków formowanych – erytrocytów, leukocytów, płytek krwi. Ich łączny efekt determinuje ciśnienie osmotyczne płynów biologicznych. Ciśnienie osmotyczne krwi ludzkiej w temperaturze 310°K (37°C) wynosi 780 kPa (7,7 atm). Takie samo ciśnienie wytwarza 0,9% wodny roztwór NaCl (0,15 mol/l), który w związku z tym jest izotoniczny z krwią (sól fizjologiczna). Jednak oprócz jonów Na i C1 we krwi znajdują się inne jony, a także wkładki i uformowane elementy. Dlatego do celów medycznych bardziej poprawne jest stosowanie roztworów zawierających te same składniki i w takiej samej ilości, jak te, które składają się na krew. Roztwory te są stosowane jako substytuty krwi w chirurgii. Organizm ludzki oprócz ciśnienia osmotycznego charakteryzuje się stałością (homeostazą) oraz innymi wskaźnikami fizykochemicznymi krwi, takimi jak kwasowość. Dopuszczalne wahania ciśnienia osmotycznego krwi są bardzo małe i nawet w ciężkiej patologii nie przekraczają kilkudziesięciu kPa. W różnych zabiegach do krwi ludzi i zwierząt można wstrzykiwać w dużych ilościach tylko roztwory izotoniczne. Przy dużych stratach krwi (na przykład po poważnych operacjach, urazach) pacjentom wstrzykuje się kilka litrów roztworu izotonicznego, aby zrekompensować utratę płynu krwią. Zjawisko osmozy znajduje szerokie zastosowanie w praktyce medycznej. Tak więc w chirurgii stosuje się opatrunki hipertoniczne (gazę nasączoną hipertonicznym 10% roztworem NaCl), które wstrzykuje się w rany ropne. Zgodnie z prawem osmozy przepływ płynu z rany przez gazę jest skierowany na zewnątrz, w wyniku czego rana jest stale oczyszczana z ropy, mikroorganizmów i produktów rozpadu. 20. Stopień dysocjacji (jonizacji). Siła elektrolitów Elektrolity, które prawie całkowicie dysocjują na jony (jonizują), nazywane są mocnymi, a elektrolity, które nie ulegają całkowitej jonizacji, nazywane są słabymi. W roztworze słabych elektrolitów wraz z jonami znajdują się niezjonizowane cząsteczki. To przez niepełną jonizację S. Arrhenius wyjaśnił, dlaczego współczynnik izotoniczny roztworów słabych elektrolitów nie jest równy liczbie całkowitej. Aby ilościowo scharakteryzować kompletność dysocjacji, wprowadzono pojęcie stopnia dysocjacji (jonizacji). Stopień dysocjacji (jonizacji) elektrolitu to stosunek liczby cząsteczek rozłożonych na jony do całkowitej liczby jego cząsteczek wprowadzonych do roztworu. Innymi słowy, an jest proporcją cząsteczek elektrolitu rozłożonych na jony. Stopień dysocjacji wyrażony jest jako procent lub ułamki jednostki: αн = Nн/ Np, gdzie N to liczba cząsteczek elektrolitu rozłożonych na jony; Np liczba cząsteczek elektrolitu wprowadzonych do roztworu (rozpuszczonych). Więc dla C(CHXNUMXCOOH) = 0,1 mol/l, stopień dysocjacji αн = 0,013 (lub 1,3%). W zależności od stopnia dysocjacji elektrolity są konwencjonalnie podzielone na silne (αн > 30%) i słabe (αн < 3%). W przedziale uważa się, że elektrolity mają średnią moc. Prawie wszystkie sole są uważane za mocne elektrolity. Z najważniejszych kwasów i zasad H2SO4 , HCI, HBr, HI, HNO3 , NaOH, KOH, Ba(OH)2 . Większość kwasów organicznych, a także niektóre związki nieorganiczne, należą do słabych elektrolitów - H2S, HCN, N2Z3 , WIĘC3 , HCl0, N2NA3W3 ,Hg2CI2 ,Fe(SCN)3 . An określa się eksperymentalnie, mierząc odchylenie właściwości porównawczych roztworów elektrolitów od teoretycznych zależności dla idealnych roztworów. Na przykład współczynnik izotoniczny i określa się metodą krioskopową, a następnie oblicza się stopień dysocjacji W przypadku silnych elektrolitów stopień dysocjacji jest widoczny, ponieważ prawie całkowicie dysocjują na jony. Odchylenie współczynnika izotonicznego i od wiedzy o liczbach całkowitych tłumaczy się im nie obecnością niezwiązanych cząsteczek w roztworze, ale innymi przyczynami. Dysocjacji towarzyszy wydzielanie lub absorpcja ciepła. Dlatego stopień dysocjacji musi zależeć od temperatury. Wpływ temperatury można oszacować zgodnie z zasadą Le Chateliera. Jeśli dysocjacja elektrolityczna postępuje z pochłanianiem ciepła, to wraz ze wzrostem temperatury wzrasta, jeśli wraz z wydzielaniem ciepła maleje. Na stopień dysocjacji elektrolitycznej wpływa stężenie roztworu. Gdy roztwór jest rozcieńczany, stopień dysocjacji znacznie wzrasta. W związku z tym wskazana klasyfikacja wytrzymałości elektrolitów według stopnia dysocjacji αн ważne tylko dla roztworów o stężeniu około 0,1 mol/l. Jeśli rozważymy dysocjację elektrolityczną jako równowagowy odwracalny proces, to zgodnie z zasadą Le Chateliera rozcieńczenie wodą zwiększa liczbę destylowanych cząsteczek, tj. zwiększa się stopień dysocjacji. Na stopień dysocjacji słabych elektrolitów wpływa również dodatek jonów o tej samej nazwie. Zatem wprowadzenie do układu równowagi słabego elektrolitu zwiększa stężenie jonów, co zgodnie z zasadą Le Chateliera prowadzi do znacznego przesunięcia równowagi dysocjacji w lewo, czyli zmniejszenia stopnia dysocjacji . Tak więc dodanie jonów o tej samej nazwie do słabego roztworu elektrolitu zmniejsza stopień jego dysocjacji. 21. Stała dysocjacji. Prawo hodowlane Ostwalda. Teoria roztworów mocnych elektrolitów Ilościowo, dysocjacja elektrolityczna jako odwracalny proces równowagowy można scharakteryzować stałą dysocjacji (jonizacji) określoną przez prawo działania mas. Ściśle rzecz biorąc, prawo działania mas dotyczy reakcji odwracalnych, czyli roztworów słabych elektrolitów. Na przykład dysocjację elektrolitu KtnAnm można przedstawić jako proces równowagowy: Ktn Anm × nKtm+ +mAn . Zgodnie z prawem działania mas, stała równowagi jest zapisana w następujący sposób: КД = (Ktm+)n+(Ann)m + (KtnAnm) gdzie (Ktm+) i (Ann ) - stężenia w równowadze molowej jonów elektrolitów; (KtnAnm) jest molowym stężeniem równowagowym niezdysocjowanych cząsteczek elektrolitu; КД jest stałą równowagi, zwaną stałą dysocjacji. To równanie obowiązuje tylko dla rozcieńczonych roztworów słabych elektrolitów. Stosując go do stężonych roztworów i do roztworów mocnych elektrolitów, równanie musi zostać zmodyfikowane. Im większa stała dysocjacji KД , tym bardziej dysocjuje elektrolit. W przeciwieństwie do stopnia dysocjacji KД zależy tylko od rodzaju rozpuszczalnika, elektrolitu i temperatury, ale nie zależy od stężenia roztworu. Zatem zarówno stała, jak i stopień dysocjacji elektrolitycznej są ilościowymi cechami dysocjacji. Naturalnie istnieje między nimi połączenie. Kwasy wielozasadowe i zasady polikwasowe dysocjują stopniowo. Na przykład dysocjacja kwasu fosforowego zachodzi w trzech etapach:  Podobnie dla zasad polikwasowych (na przykład Ca (OH)2) - dysocjacja zachodzi w dwóch etapach. Dysocjacja stopniowa charakteryzuje się tym, że rozkład elektrolitu na każdym kolejnym etapie zachodzi w mniejszym stopniu niż na poprzednim. Taką zmianę stałych dysocjacji można wyjaśnić przyciąganiem elektrostatycznym na podstawie prawa Coulomba. Energia jonizacji jest minimalna, gdy jon zostaje odłączony od cząsteczki obojętnego elektrolitu. Oderwanie jonu na każdym kolejnym etapie dysocjacji wymaga coraz większej energii, ponieważ usuwanie jonu następuje z cząstki, której ładunek zwiększa się w kolejnych etapach. Prawie całkowitą dysocjację mocnych elektrolitów na jony, niezależnie od stężenia ich roztworów, potwierdzają metody badań fizycznych i fizykochemicznych. Zatem wartości ciepła neutralizacji wszystkich mocnych kwasów przez mocne zasady w rozcieńczonych roztworach są prawie takie same. Niezależnie od charakteru kwasu i zasady otrzymuje się tę samą wartość AH = 56,5 kJ/mol. Fakt ten jest wyraźnym dowodem całkowitej dysocjacji rozcieńczonych roztworów kwasów i zasad. We wszystkich przypadkach powszechnym procesem zachodzącym podczas neutralizacji jest połączenie jonów w molu 22. Teoria kwasów i zasad Wiele elektrolitów, w szczególności wodorotlenki różnych pierwiastków E, wykazuje właściwości kwasów lub zasad. Dysocjacja wodorotlenku EON może przebiegać w dwóch rodzajach: 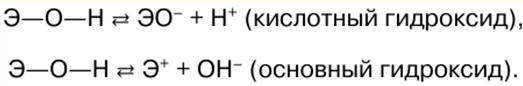 Luka może wystąpić wzdłuż obu wiązań grupy WIECZNOŚĆ. Jak wiadomo, polarność i siła wiązań zależą od różnicy elektroujemności pierwiastków, wielkości i efektywnego ładunku atomów. Jeżeli energia rozerwania wiązania O-H jest znacznie mniejsza niż energia rozerwania wiązania E-O, to dysocjacja wodorotlenku przebiega zgodnie z typem kwasu. Jeśli wręcz przeciwnie, energia zerwania wiązania O-H jest znacznie większa niż energia zerwania wiązania E-O, to dysocjacja przebiega zgodnie z głównym typem. W wodorotlenkach metali alkalicznych i metali ziem alkalicznych, a także metali przejściowych na niskich stopniach utlenienia siła wiązania E-O jest stosunkowo niska, tlen jest silniej związany z wodorem, a dysocjacja E-O-H przebiega głównie według typu podstawowego, tj. z eliminacją wodorotlenku dionu. Wynika to z faktu, że jony takich pierwiastków są dość duże i mają mały ładunek efektywny, tj. mają słabą zdolność polaryzacyjną. Wraz ze wzrostem stopnia utlenienia wzrasta działanie polaryzacyjne atomu E (zwiększa się ładunek właściwy), tlen jest mocniej związany z pierwiastkiem E, a dysocjacja E-O-H zachodzi głównie w zależności od rodzaju kwasu, tj. jonu wodorowego jest oddzielony. To ostatnie wiąże się z redystrybucją gęstości elektronów na atomie tlenu. W rezultacie wiązanie E-O staje się silniejsze, a wiązanie OH słabnie. Obecnie nie ma jednoznacznej definicji pojęć kwas i zasada, którą można by w równym stopniu wykorzystać do scharakteryzowania oddziaływań kwasowo-zasadowych w dowolnych rozpuszczalnikach. Do scharakteryzowania wielu elektrolitów w roztworach wodnych nadal można stosować pojęcia kwasu i zasady podane przez Arrheniusa: 1) kwas jest elektrolitem, który dysocjuje w roztworach, tworząc jony wodorowe H; 2) zasada jest elektrolitem, który dysocjuje w roztworach z utworzeniem jonów wodorotlenowych OH; 3) amfolit (wodorotlenek amfoteryczny) jest elektrolitem, który dysocjuje w roztworze, tworząc zarówno jony wodorowe, jak i jony wodorotlenkowe. Amfolity obejmują wodorotlenki cynku, glinu, chromu i innych pierwiastków amfoterycznych, a także aminokwasy, białka, kwasy nukleinowe. Zastosowanie zasady Le Chateliera do łańcucha równowag kwasowo-zasadowych pokazuje, że wraz ze wzrostem stężenia hydroksydionów OH w układzie wzrasta prawdopodobieństwo dysocjacji typu kwasowego. Wzrost stężenia jonów wodorowych H+ w układzie prowadzi do preferencyjnej dysocjacji według głównego typu. Oznacza to, że w środowisku kwaśnym amfolit ma charakter zasadowy, a w środowisku zasadowym kwaśny. Na przykład wodorotlenek cynku zachowuje się jak zasada podczas interakcji z kwasami: Zn (OH)2 + 2HCl - ZnCI2 + 2H2Och a podczas interakcji z alkaliami - jako kwas: Zn(OH)2+ 2NaOH → Na2[Zn(OH)4]. 23. Systemy buforowe krwi Plazma krwi Systemy buforowe mają ogromne znaczenie w utrzymaniu równowagi kwasowo-zasadowej organizmów. Płyny wewnątrzkomórkowe i zewnątrzkomórkowe wszystkich żywych organizmów charakteryzują się stałą wartością pH, utrzymywaną za pomocą systemów buforowych. Wartość pH większości płynów wewnątrzkomórkowych mieści się w zakresie od 6,8 do 7,8. Równowagę kwasowo-zasadową bilansu CO w ludzkiej krwi zapewniają układy buforowe wodorowęglanowe, fosforanowe i białkowe. Prawidłowa wartość pH osocza krwi wynosi 7,40 ± 0. Odpowiada to zakresowi wartości kwasowości czynnej od 05 do 3,7x4,0 mol/l. Ponieważ we krwi znajdują się różne elektrolity (HC1083 H2CO3 , H2RO4 , NRO42 ), białka, aminokwasy, co oznacza, że dysocjują do tego stopnia, że aktywność a (H+) mieści się we wskazanym zakresie. Ze względu na to, że zawartość substancji nieorganicznych i organicznych w osoczu i komórkach krwi nie jest taka sama, wskazane jest oddzielne rozpatrywanie tych składników krwi. Osocze krwi System buforowy HCO3 / N2Z3 składa się z kwasu węglowego2Z3 i sprzężona zasada HCO3 . Jest to najważniejszy układ buforowy krwi. Jednym ze składników jest kwas węglowy H2Z3 - powstaje w wyniku oddziaływania CO rozpuszczonego w plazmie2 z wodą: Z2(r) + H2On N2Z3. gdzie CO2(r) - stężenie rozpuszczonego CO2 . Stała równowagi tej reakcji to: K = [N2Z3] / [CO2] Między CO2 w pęcherzykach płucnych i buforze wodorowęglanowym w osoczu krwi przepływającym przez naczynia włosowate płuc ustala się łańcuch równowagi. System buforowy wodorowęglanu działa jako skuteczny bufor fizjologiczny w pobliżu pH 7,4. Po wejściu do krwi kwasów - dawcy H+ równowaga w łańcuchu zgodnie z zasadą Le Chateliera przesuwa się w lewo w wyniku tego, że jony HCO3 wiążą jony H w cząsteczki H2Z3 . Jednocześnie stężenie H2Z3 wzrasta, a stężenie jonów HCO3 idzie w dół. Zwiększenie stężenia H2Z3 prowadzi do przesunięcia równowagi w lewo (zasada Le Chateliera). Powoduje rozkład2Z3 i wzrost stężenia CO2 rozpuszczony w osoczu. W rezultacie równowaga przesuwa się w lewo i wzrasta ciśnienie CO.2 w płucach. Nadmiar CO2 wydalany z organizmu. W rezultacie układ wodorowęglanowy krwi szybko osiąga równowagę z CO2 w pęcherzykach płucnych i skutecznie utrzymuje stałe pH osocza krwi. 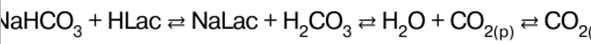 W ten sposób utrzymywana jest normalna wartość pH krwi z nieznacznie wyraźną zmianą pH spowodowaną kwasicą. W zamkniętych pomieszczeniach często doświadczają uduszenia (braku tlenu), wzmożonego oddychania. Jednak uduszenie wiąże się nie tyle z brakiem tlenu, ile z nadmiarem CO2. Nadmiar CO2 w atmosferze, zgodnie z prawem Henry'ego, prowadzi do dodatkowego rozpuszczenia CO2 we krwi. A to prowadzi do obniżenia pH krwi, czyli do kwasicy. System buforowy wodorowęglanowy najszybciej reaguje na zmiany pH krwi. Jego pojemność buforowa kwasu to Vк \u40d XNUMX mmol / l osocza krwi, a pojemność buforowa dla zasad jest znacznie mniejsza i jest w przybliżeniu równa Vщ = 1-2 mmol/l osocza krwi. 24. Reakcje neutralizacji Reakcje neutralizacji nazywane są reakcjami wymiany interakcji kwasów i zasad, w wyniku których powstaje sól i woda. Rozważ różne rodzaje reakcji neutralizacji. 1. Neutralizacja mocnej zasady mocnym kwasem: KOH + HNO3 -KNO3 + H2O. Równanie jonów cząsteczkowych dla takiej reakcji H+ + OH → N2O a ujemna wartość energii Gibbsa ΔG° pokazuje, że równowaga jest praktycznie przesunięta w kierunku tworzenia się wody. Częstym przypadkiem reakcji neutralizacji jest oddziaływanie kwasów i zasad różniących się siłą (stopień dysocjacji). Reakcje te nie dobiegają końca ze względu na odwrotną reakcję hydrolizy soli. 2. Neutralizacja słabego kwasu mocną zasadą:  lub w postaci molekularnej:  W tym przypadku reakcja neutralizacji jest odwracalna. Reakcja neutralizacji słabej zasady mocnym kwasem jest również odwracalna:  lub w postaci molekularnej: 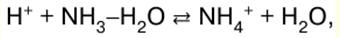 a także - słaba zasada ze słabym kwasem: 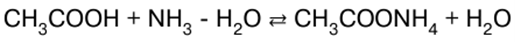 lub w postaci molekularnej:  W tych układach równowaga jest silnie przesunięta w prawo, ponieważ woda jest znacznie słabszym elektrolitem niż kwas cyjanowodorowy, amoniak i kwas octowy. Podstawą metody neutralizacji są reakcje neutralizacji. Ta metoda jest stosowana w laboratoriach klinicznych do określania kwasowości soku żołądkowego, pojemności buforowej osocza krwi. W farmakologii służy do ilościowej analizy kwasów nieorganicznych (chlorowodorowy, siarkowy, borowy) i organicznych (octowy, benzoesowy, winowy, cytrynowy, salicylowy). W badaniach biofarmaceutycznych pKa kwasów i pKa zasad określa się przez neutralizację, ponieważ wartość tych wartości może przewidywać zdolność leków do przechodzenia przez błony biologiczne. Do wyznaczenia pK . stosuje się miareczkowanie kwasowo-zasadoweа aminokwasy i pKа grupy dysocjujące w białkach. Krzywe miareczkowania białek otrzymane w dwóch różnych temperaturach można wykorzystać do określenia liczby grup karboksylowych, imidazolowych i innych. Miareczkowanie aminokwasów i białek umożliwia wyznaczenie ich punktów izoelektrycznych. Hydroliza to rozkład substancji przez wodę. Hydrolizie mogą ulegać związki chemiczne różnych klas: białka, tłuszcze, węglowodany, estry, sole itp. W chemii nieorganicznej najczęściej spotyka się je z hydrolizą soli. 25. Hydroliza soli Hydroliza solna - jest to oddziaływanie soli z cząsteczkami wody, prowadzące do powstania związków o niskiej dysocjacji. Proces hydrolizy polega na przejściu protonu z cząsteczki wody do danego jonu (CO32 + HOH * HCO3+ OH ) lub z danego jonu, w tym z uwodnionego kationu metalu, do cząsteczki wody. W zależności od charakteru soli, woda działa jako kwas lub jako zasada, a sól jest odpowiednio sprzężoną zasadą lub sprzężonym kwasem. W zależności od rodzaju soli możliwe są cztery warianty hydrolizy. 1. Sole utworzone przez mocny kwas i słabą zasadę:  2. Sole utworzone przez mocną zasadę i słaby kwas: CH3COONa + HOH → CH3COOH + NaOH. 3. Sole utworzone przez słaby kwas i słabą zasadę. Cyjanek amonu hydrolizuje w reakcji: 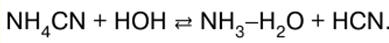 4. Sole utworzone przez mocny kwas i mocną zasadę. NaCl, KNO3 nie ulegają hydrolizie. Mechanizm hydrolizy soli polega na polaryzacyjnym oddziaływaniu jonów soli z ich powłoką hydratacyjną. Im silniejsze to oddziaływanie, tym intensywniejsza hydroliza. Wszystkie rozważane przypadki hydrolizy dotyczyły soli tworzonych przez zasady jednokwasowe i kwasy jednozasadowe. Sole kwasów wielozasadowych i zasad wielokwasowych hydrolizuje się stopniowo, tworząc sole kwasowe i zasadowe. Ilościowo hydroliza jako równowaga CO charakteryzuje się stopniem hydrolizy i stałą hydrolizy. Stopień hydrolizy mierzy się stosunkiem ilości zhydrolizowanej substancji do całkowitej ilości substancji rozpuszczonej. Stopień hydrolizy zależy od charakteru soli, jej stężenia i temperatury. Zgodnie z prawem działania masy stopień hydrolizy wzrasta wraz z rozcieńczeniem roztworu. Tak więc w koncentracji Na2CO3 0,001 mol/l, stopień hydrolizy wynosi 34%. W ogólnym przypadku prawdziwe są następujące prawidłowości. 1. Hydroliza soli powinna wzrastać wraz ze wzrostem temperatury i rozcieńczaniem roztworu. 2. W hydrolizie odwracalnej, zgodnie z zasadą Le Chateliera, proces powinien być stłumiony przez zakwaszenie (jeśli ta sól jest tworzona przez mocny kwas i słabą zasadę, jony H kumulują się) lub przez alkalizację (jeśli sól jest tworzona przez słaby kwas i mocna zasada, jony OH gromadzą się). 3. Hydroliza soli, w wyniku której powstają słabo rozpuszczalne lub gazowe produkty usuwane ze sfery reakcyjnej (zasada przesunięcia równowagi), jest nieodwracalna. Na przykład hydroliza Pb(SO4)2 przebiega całkowicie z powodu tworzenia się osadu PbO2: Pb(SO4)2 + 2H2O→PbO2 + 2H2SO4. Hydroliza jest charakterystyczna dla wielu klas związków nieorganicznych i organicznych. Hydroliza związków nieorganicznych jest ważna dla oceny ich toksyczności. Hydroliza związków organicznych służy do pozyskiwania cennych produktów z drewna, tłuszczów, estrów i innych, jednak hydroliza odgrywa szczególnie ważną rolę w życiu organizmów żywych. 26. Wytrącanie i reakcja rozpuszczania Reakcje wymiany zachodzące w roztworze elektrolitu obejmują reakcje wytrącania i rozpuszczania. Reakcjom wytrącania towarzyszy wytrącanie. Reakcje, którym towarzyszy rozpuszczanie osadów, nazywane są reakcjami rozpuszczania. Powszechnie stosowane są układy składające się ze złoża trudno rozpuszczalnego elektrolitu i znajdującego się nad nim roztworu nasyconego. W takich układach ustala się dynamiczna równowaga między roztworem nasyconym a osadem. Ze względu na niską rozpuszczalność stężenie trudno rozpuszczalnego elektrolitu w roztworze jest bardzo niskie, dlatego można uznać, że jest on całkowicie zdysocjowany w roztworze. Innymi słowy, dynamiczna równowaga w nasyconym roztworze ustala się między fazą stałą substancji a jonami, które przeszły do roztworu. Na przykład w nasyconym roztworze AgCl zachodzi następująca równowaga: AgCl(T) → Ag+(P) +Cl(R). Stężenie fazy stałej AgCl jako wartość stała jest wykluczone z wyrażenia na stałą równowagi. W rezultacie stała równowagi jest określana tylko przez iloczyn stężeń jonów w roztworze i nazywana jest stałą lub iloczynem rozpuszczalności. W ogólnym przypadku dla elektrolitu Ktn Anm stałą rozpuszczalności określa iloczyn stechiometryczny stężeń jonów: Кпр= [Ktm+ ]n[Jakiśn]m Ta wartość charakteryzuje rozpuszczalność elektrolitu w stałej temperaturze przy braku obcych substancji. Stałość Kпр nie oznacza stałości stężeń poszczególnych jonów w roztworze. Dzięki temu możliwe jest zwiększenie stężenia jonów Ag w nasyconym roztworze AgCl poprzez dodanie np. AgNO3 , podczas gdy równowaga zgodnie z zasadą Le Chateliera przesunie się w lewo, co doprowadzi do wzrostu szybkości osadzania jonów. Po pewnym czasie szybkości rozpuszczania AgCl i wytrącania jonów Ag i Cl zrównają się. Nowo ustanowioną równowagę, jak poprzednio, będzie charakteryzować wartość Kпр(AgCl), ale stężenia równowagowe jonów Ag i Cl ulegną zmianie. Tak więc, w oparciu o Kпр możliwe jest przewidzenie powstawania i rozpuszczania wydzieleń elektrolitu na podstawie dwóch reguł. 1. Elektrolit wytrąca się, gdy iloczyn stechiometryczny stężeń jego jonów w roztworze jest większy niż stała rozpuszczalności. 2. Osad elektrolitu rozpuszcza się, gdy iloczyn stechiometryczny stężeń jego składowych jonów w roztworze staje się mniejszy niż stała rozpuszczalności. Reakcje wytrącania stanowią podstawę metody wytrącania, którą wykorzystuje się w analizie ilościowej farmaceutyków. Metodę strąceniową stosuje się w analizie klinicznej chlorków w moczu, soku żołądkowym, krwi oraz w praktyce sanitarno-higienicznej – w analizie wody pitnej. Naukowcy uważają, że różna rozpuszczalność naturalnych związków pierwiastków w wodzie ma ogromny wpływ na ich zawartość w organizmach żywych. Istnieje ścisły związek pomiędzy rozpuszczalnością związków w wodzie a toksycznym działaniem jonów szeregu pierwiastków. Przykładowo wprowadzenie Al3 + do organizmu ze względu na powstawanie słabo rozpuszczalnego fosforanu glinu AlPO4 27. Reakcje redoks Jednym z podstawowych pojęć chemii nieorganicznej jest pojęcie stopnia utlenienia (CO). Stan utlenienia pierwiastka w związku to formalny ładunek atomu pierwiastka, obliczony z założenia, że elektrony walencyjne przechodzą do atomów o wyższej względnej elektroujemności (REO), a wszystkie wiązania w cząsteczce związku są jonowe. Stopień utlenienia pierwiastka E jest oznaczony na górze symbolu pierwiastka znakiem „+” lub „-” przed liczbą. Stopień utlenienia jonów faktycznie występujących w roztworze lub kryształach pokrywa się z ich liczbą ładunku i podobnie jest oznaczony znakiem „+” lub „-” po liczbie, np. Cl,Ca2+. Metodę Stock stosuje się również do oznaczenia stopnia utlenienia cyframi rzymskimi po symbolu pierwiastka: Mn (VII), Fe (III). Kwestię znaku stopnia utlenienia atomów w cząsteczce rozwiązuje się na podstawie porównania elektroujemności połączonych ze sobą atomów tworzących cząsteczkę. W tym przypadku atom o niższej elektroujemności ma dodatni stopień utlenienia, a atom o wyższej elektroujemności ma ujemny stopień utlenienia. Należy zauważyć, że stopnia utlenienia nie można utożsamiać z wartościowością pierwiastka. Wartościowość, definiowana jako liczba wiązań chemicznych, którymi dany atom jest połączony z innymi atomami, nie może być równa zeru i nie posiada znaku „+” ani „-”. Stopień utlenienia może mieć zarówno wartość dodatnią, jak i ujemną, ale może również przyjmować wartość zerową lub nawet ułamkową. A więc w cząsteczce CO2 stopień utlenienia C wynosi +4, a w cząsteczce CH4 stopień utlenienia C wynosi 4. Wartościowość węgla4 a w obu związkach wynosi IV. Pomimo powyższych wad, stosowanie pojęcia stopnia utlenienia jest wygodne w klasyfikacji związków chemicznych i formułowaniu równań dla reakcji redoks. Gdy pierwiastek jest utleniony, stopień utlenienia wzrasta, innymi słowy, czynnik redukujący podczas reakcji zwiększa stopień utlenienia. Wręcz przeciwnie, gdy pierwiastek jest redukowany, zmniejsza się stopień utlenienia, tj. podczas reakcji środek utleniający zmniejsza stopień utlenienia. W ten sposób można podać następujące sformułowanie reakcji redoks: reakcje redoks to reakcje zachodzące ze zmianą stopnia utlenienia atomów pierwiastków tworzących substancje reagujące. 28. Środki utleniające i redukujące Aby przewidzieć produkty i kierunek reakcji redoks, warto pamiętać, że typowymi utleniaczami są proste substancje, których atomy mają duże EER >3,0 (elementy grup VIA i VIIA). Spośród nich najsilniejszymi utleniaczami są fluor (OEO = 4,0), tlen (OEO = 3,0), chlor (OEO = 3,5). Ważne utleniacze obejmują PbO2 , KMnO4 , Ca(SO4)2 , K.2Cr2O7 , HClO, HClO3KSIO4, NaBio3H2SO4(koniec), H.N.O.3(koniec) , Na2O2 , (NH4)2S2O8 KSIO3 H2O2 oraz inne substancje, które zawierają wyższe lub wyższe atomy CO. Typowymi środkami redukującymi są proste substancje, których atomy mają małe EER < 1,5 (metale z grup IA i IIA oraz niektóre inne metale). Ważnymi czynnikami redukującymi są H2S, N.H.3, HI, KI, SnCI2 , FeSOXNUMX4 ,C,H2 ,CO,H2SO3 , cr2(SO4)3 , CuCI, Na2S2O3 i inne substancje, które zawierają niskie atomy CO. Substancje zawierające atomy odpowiednio na maksymalnym i minimalnym stopniu utlenienia mogą być tylko środkami utleniającymi, na przykład K2SG2O7 , KMPO4 , PBO2 , HClO4 lub tylko środki redukujące, takie jak NH3 H2S, CZEŚĆ. Substancje zawierające atomy na pośrednich stanach utlenienia są zdolne zarówno do podnoszenia, jak i obniżania stanu utlenienia, to znaczy mogą być zarówno środkami redukującymi (pod działaniem bardziej aktywnego środka utleniającego niż one), jak i środkami utleniającymi (pod działaniem bardziej aktywny niż one , środek redukujący). Takie substancje wykazują dualizm redoks. Przy zestawieniu równań reakcji redoks można zastosować dwie metody: metodę równowagi elektronowej i metodę jonowo-elektroniczną (metoda półreakcji). Bardziej poprawne wyobrażenie o procesach redoks w roztworach zapewnia metoda jonowo-elektroniczna. Dzięki tej metodzie zmiany, które faktycznie zachodzą w roztworze, są przewidywane na podstawie jonów i cząsteczek. Oprócz przewidywania produktów reakcji, jonowe równania połówkowej reakcji są niezbędne do zrozumienia procesów redoks zachodzących podczas elektrolizy oraz w ogniwach galwanicznych. Metoda ta odzwierciedla rolę środowiska jako uczestnika procesu. I wreszcie, stosując tę metodę, nie trzeba z góry znać wszystkich utworzonych substancji, ponieważ wiele z nich uzyskuje się przez zestawienie równania reakcji redoks. Należy pamiętać, że choć połówkowe reakcje odzwierciedlają rzeczywiste procesy zachodzące podczas reakcji redoks, to nie można ich utożsamiać z rzeczywistymi etapami (mechanizmami) reakcji redoks. Na charakter i kierunek reakcji redoks wpływa wiele czynników: rodzaj reagentów, reakcja ośrodka, stężenie, temperatura i katalizatory. Należy pamiętać, że wartość ujemna nie zawsze prowadzi do jednoznacznej decyzji o rzeczywistym przebiegu reakcji w danym kierunku, gdyż dodatkowo konieczne jest uwzględnienie czynnika kinetycznego. 29. Biologiczne znaczenie procesów redoks Reakcje redoks to procesy chemiczne, którym towarzyszy przenoszenie elektronów z jednej cząsteczki lub jonu na drugą. W reakcjach redoks zachodzą dwa powiązane ze sobą procesy: utleniania i redukcji. Utlenianie to proces utraty elektronów. Odzyskiwanie to proces dodawania elektronów. Substancje, których atomy lub jony oddają elektrony, nazywane są czynnikami redukującymi. Substancje, których atomy lub jony dodają elektrony (lub przyciągają do siebie wspólną parę elektronów), nazywane są środkami utleniającymi. W reakcji cynku z CuSO4 Cu2 + dodaj elektrony: sami2+ + 2mi- - Si0 . Atomy cynku oddają elektrony: Zn0 -Zn2 + 2mi-. W związku z tym CuSO4 - środek utleniający, Zn - środek redukujący. Ważnymi procesami zachodzącymi w organizmach zwierzęcych są reakcje enzymatycznego utleniania substancji substratowych: węglowodanów, tłuszczów, aminokwasów. W wyniku tych procesów organizmy otrzymują duże ilości energii. Około 90% całkowitego zapotrzebowania energetycznego dorosłego mężczyzny pokrywana jest przez energię wytwarzaną w tkankach w wyniku utleniania węglowodanów i tłuszczów. Pozostała część energii – ~10% – pochodzi z oksydacyjnego rozkładu aminokwasów. Utlenianie biologiczne przebiega przez złożone mechanizmy z udziałem dużej liczby enzymów. W mitochondriach utlenianie zachodzi w wyniku przeniesienia elektronów z substratów organicznych. Jako nośniki elektronów łańcuch oddechowy mitochondriów zawiera różne białka zawierające różne grupy funkcyjne, które są przeznaczone do przenoszenia elektronów. Gdy przemieszczają się wzdłuż łańcucha od jednego pośrednika do drugiego, elektrony tracą swoją energię swobodną. Na każdą parę elektronów przeniesionych przez łańcuch oddechowy do tlenu syntetyzowane są 3 cząsteczki ATP. Energia swobodna uwalniana podczas transferu 2 elektronów do tlenu wynosi 220 kJ/mol. Synteza 1 cząsteczki ATP w standardowych warunkach zużywa 30,5 kJ. Z tego jasno wynika, że dość znaczna część energii swobodnej uwalnianej podczas przenoszenia jednej pary elektronów jest magazynowana w cząsteczkach ATP. Z tych danych jasno wynika również rola wieloetapowego przeniesienia elektronów z początkowego czynnika redukującego do tlenu. Duża energia (220 kJ) uwalniana podczas przenoszenia jednej pary elektronów do tlenu jest dzielona na szereg części odpowiadających poszczególnym etapom utleniania. Na trzech takich etapach ilość uwolnionej energii w przybliżeniu odpowiada energii wymaganej do syntezy 1 cząsteczki ATP. Reakcje redoks stanowią podstawę metod oksydymetrycznych, które w analizie klinicznej wykorzystywane są do oznaczania jonów Ca, kwasu moczowego, enzymów katalazy i peroksydazy, cukru we krwi, a w analizach sanitarno-higienicznych – do oznaczania utlenialności wody, zawartości aktywny chlor w wybielaczu, chlor resztkowy w wodzie pitnej w gospodarstwie domowym 30. Wiązanie chemiczne i jego charakterystyka eksperymentalna Opracowanie nowoczesnego modelu atomu i przewidywanie na jego podstawie właściwości poszczególnych atomów jest bardzo ważnym osiągnięciem mechaniki kwantowej. Jednak izolowane atomy rzadko można znaleźć w warunkach ziemskich. Otaczające nas ciała przyrody nieożywionej i żywej składają się z różnorodnych cząsteczek. A. M. Butlerov (1828-1886) stworzył teorię struktury chemicznej substancji organicznych (1861). Od tego czasu pojęcia „wartościowości” i „wiązania chemicznego” stopniowo zaczęły wkraczać do chemii. Walencja to zdolność atomu do przyłączenia pewnej liczby innych atomów w celu utworzenia cząsteczki. Wartościowość jest oznaczona myślnikami obok symbolu pierwiastka. Wodór (H) jest jednowartościowy, tlen (0=) jest dwuwartościowy. Liczba słupków walencyjnych określa liczbę wiązań chemicznych, jakie dany atom może utworzyć z innymi atomami. Wiązanie chemiczne to zestaw interakcji między elektronami a jądrami, prowadzący do połączenia atomów w cząsteczkę. Właściwości wiązania chemicznego bada się różnymi metodami. Za pomocą metod chemicznych określa się liczbę wiązań atomów (wartościowość) i ich reaktywność. Za pomocą metod fizycznych określa się długość, siłę, orientację i polarność wiązań chemicznych. Długość wiązania chemicznego rс nazywana wartością mierzoną odległością między jądrami związanych atomów. Jako jednostka długości wiązania chemicznego gс wygodny w użyciu pikometr (pm): 1:10 = XNUMX12 m Siła wiązania chemicznego Eс - wartość mierzona entalpią ΔЕс tworzenie połączenia. Jako jednostka siły wiązania chemicznego Eс stosuje się kJ/mol. Orientacja wiązania chemicznego aс - wartość mierzona kątem między kierunkami wiązań danego atomu z sąsiednimi atomami cząsteczki. Kąt ac nazywamy kątem walencyjnym. Jednostka kąta wiązania aс - stopień. Polaryzacja wiązania chemicznego μс - wartość mierzona momentem elektrycznym tego połączenia. Moment elektryczny dwóch ładunków elektrycznych +q i ־q, równy wartości bezwzględnej i przeciwny znak, jest równy μ = qr, gdzie r jest odległością między ładunkami. Te dwa ładunki tworzą dipol elektryczny. Wiązanie chemiczne jest spolaryzowane, gdy 2 atomy o różnej elektroujemności (EOE) wiążą się. W rezultacie na atomie o dużej wartości OEO pojawia się nadmierny ładunek ujemny δ, a na innym atomie o niższej wartości OEO pojawia się nadmiar ładunku dodatniego +δ. Polaryzację połączenia oblicza się ze wzoru: μс = rс. Wygodnie jest używać niesystemowej jednostki Debye'a (D) jako jednostki do pomiaru polarności wiązania chemicznego - 1 D = 3,3 x 1030 C/m. Polaryzacja wiązania OH w cząsteczce wody wynosi μon = 1,5D. Badanie wiązania chemicznego wykazało, że w większości przypadków długość, wytrzymałość, orientacja, polarność tego samego wiązania chemicznego w różnych związkach mają w przybliżeniu takie same wartości. Wynika stąd, że oddziaływania prowadzące do powstania danego wiązania między atomami mają ten sam charakter w różnych cząsteczkach. Teorie mechaniki kwantowej wiązania chemicznego wyjaśniają ten fakt. 31. Wiązanie wodorowe. Międzycząsteczkowe i wewnątrzcząsteczkowe wiązania wodorowe Wiązania chemiczne w cząsteczkach są zwykle bardzo silne, ich energia mieści się w zakresie 100-150 kJ/mol. Ponadto istnieją tak zwane wiązania wodorowe, których siła wynosi 10-40 kJ/mol. Długość tych wiązań wynosi odpowiednio 270-230 pm. Wiązanie wodorowe między atomami EА i EВ nazwane oddziaływaniem przeprowadzanym przez atom wodoru połączony z EА kruszecВ wiązanie chemiczne. Obraz wiązania wodorowego w ogólnym przypadku ma postać: ЭА-N...Eв.. Oczywiście wiązanie wodorowe jest trzycentryczne, ponieważ w jego tworzeniu biorą udział 3 atomy. Do wystąpienia takiego wiązania konieczne jest, aby atomy EА i EВ mają wysoką elektroujemność. Są to atomy najbardziej negatywnych pierwiastków: azotu (REO = 3,0), tlenu (REO = 3,5), fluoru (REO = 4,0) i chloru (REO = 3,0). Wiązanie wodorowe powstaje w wyniku połączenia wodoru ls-AO i dwóch atomów 2pAO EА i EВ; 2 orbitale są zorientowane wzdłuż jednej linii prostej. Dlatego wiązanie wodorowe jest liniowe. Wiązanie wodorowe nazywa się: 1) wewnątrzcząsteczkowy, jeśli atomy EА i EВ , połączone tym wiązaniem, należą do tej samej cząsteczki; 2) międzycząsteczkowy, jeśli atomy EА i EВ są w różnych cząsteczkach. Wewnątrzcząsteczkowe wiązania wodorowe odgrywają istotną rolę biologiczną, ponieważ determinują na przykład helikalną strukturę cząsteczek białek polimerowych. W białkach są to wiązania N-H...O pomiędzy resztami aminokwasów. Nie mniej ważne są międzycząsteczkowe wiązania wodorowe. Za ich pomocą łańcuchy kwasów nukleinowych łączą się, tworząc podwójną helisę. Istnieją dwa rodzaje wiązań pomiędzy zasadami nukleinowymi – NHN i N-H-O. Średnia energia kinetyczna ruchu termicznego cząsteczek jest rzędu 3/2RT. Przy temperaturze ciała człowieka wynoszącej 37°C (310°K) jest to około 4 kJ/mol. Siła wiązań wodorowych mieści się w zakresie 10-40 kJ/mol, dzięki czemu są wystarczająco silne, aby wytrzymać stałe uderzenia otaczających cząsteczek i zapewnić niezmienność kształtu polimerowych struktur biologicznych. Jednocześnie pod wpływem aktywnych cząsteczek wiązania wodorowe ulegają okresowemu zerwaniu, a następnie odbudowują się, zapewniając przepływ różnych procesów życiowych. Rozważane przykłady wyraźnie ilustrują szerszy zakres zastosowań metody MO LCAO niż metody VS. Niemniej jednak metoda VS może być z powodzeniem stosowana do przewidywania właściwości i struktury wielu substancji, w tym związków złożonych. 32. Makro i mikroelementy w środowisku i w organizmie człowieka Istnieją różne klasyfikacje pierwiastków chemicznych zawartych w ludzkim ciele. Tak więc V. I. Vernadsky, w zależności od średniej zawartości (ułamek masowy w, %) w żywych organizmach, podzielił elementy zgodnie z systemem dziesięciodniowym. Zgodnie z tą klasyfikacją pierwiastki zawarte w organizmach żywych dzielą się na trzy grupy: makro, mikro i ultramikroelementy. Makroskładniki Są to pierwiastki, których zawartość w organizmie jest wyższa niż 102%. Należą do nich tlen, węgiel, wodór, azot, fosfor, siarka, wapń, magnez, sód i chlor. Elementy śledzenia Są to pierwiastki, których zawartość w organizmie mieści się w przedziale 103 do 105%. Należą do nich jod, miedź, arsen, fluor, brom, stront, bar, kobalt. Ultramikroelementy Są to elementy, których zawartość w ciele jest poniżej 105%. Należą do nich rtęć, złoto, uran, tor, rad itp. Obecnie ultramikroelementy łączy się z mikroelementami w jedną grupę. Ta klasyfikacja odzwierciedla tylko zawartość pierwiastków w żywych organizmach, ale nie wskazuje na biologiczną rolę i fizjologiczne znaczenie tego lub innego pierwiastka. V. V. Kovalsky, w oparciu o ich znaczenie dla życia, podzielił pierwiastki chemiczne na trzy grupy. Niezbędne (niezastąpione) elementy Są stale zawarte w organizmie człowieka, wchodzą w skład enzymów, hormonów i witamin: H, O, Ca, N, K, P, Na, S, Mg, d, C, I, Mn, Cu, Co, Fe, Zn, Mo, V. Ich niedobór prowadzi do zakłócenia normalnego życia człowieka. elementy nieczystości Te pierwiastki stale znajdują się w ciele zwierząt i ludzi: Ga, Sb, Sr, Br, F, B, Be, Li, Si, Sn, Cs, Al, Ba, Ge, As, Rb, Pb, Ra, Bi, Cd, Cr, Ni, Ti, Ag, Th, Hg, U, Se. Ich biologiczna rola jest mało poznana lub nieznana. elementy nieczystości Sc, Tl, In, La, Pr, Sm, W, Re, Tb itp. Występuje u ludzi i zwierząt. Dane dotyczące liczby i roli biologicznej nie zostały jeszcze wyjaśnione. Pierwiastki niezbędne do budowy i aktywności życiowej różnych komórek i organizmów nazywane są pierwiastkami biogennymi. Wciąż nie jest możliwe dokładne wymienienie wszystkich pierwiastków biogennych ze względu na trudności w określeniu bardzo niskich stężeń pierwiastków śladowych i ustaleniu ich funkcji biologicznych. Dla 24 pierwiastków biogenność została ustalona wiarygodnie. Są to elementy pierwszej i niektóre elementy drugiej grupy (według Kowalskiego). 33. Topografia najważniejszych pierwiastków biogennych w organizmie człowieka Narządy ludzkie w różny sposób koncentrują w sobie różne pierwiastki chemiczne, tzn. mikro i makroelementy są nierównomiernie rozmieszczone w różnych narządach i tkankach. Większość pierwiastków śladowych gromadzi się w tkankach wątroby, kości i mięśni. Tkanki te są głównymi magazynami (magazynami) wielu pierwiastków śladowych. Pierwiastki mogą wykazywać specyficzne powinowactwo do określonych narządów i występować w nich w wysokich stężeniach. Powszechnie wiadomo, że cynk koncentruje się w trzustce, jod w tarczycy, fluor w szkliwie zębów, glin, arsen, wanad gromadzą się we włosach i paznokciach, kadm, rtęć, molibden w nerkach, cyna w jelitach. tkanki, stront – w gruczole krokowym, tkance kostnej, bar – w siatkówce barwnikowej, brom, mangan, chrom – w przysadce mózgowej itp. W organizmach mikroelementy mogą występować w stanie związanym oraz w postaci wolnych form jonowych. Wiadomo, że krzem, glin, miedź i tytan w tkance mózgowej występują w postaci kompleksów z białkami, natomiast mangan występuje w formie jonowej. Wodór i tlen to makroelementy. Wchodzą w skład wody, która w organizmie dorosłego człowieka stanowi średnio około 65%. Woda jest nierównomiernie rozprowadzana w ludzkich narządach, tkankach i płynach biologicznych. Tak więc w soku żołądkowym, ślinie, osoczu krwi, limfie, wodzie stanowi od 89,5 do 90%, w moczu, istocie szarej mózgu, nerkach - 80%, w istocie białej mózgu, wątrobie, skórze, rdzeniu kręgowym , mięśnie, płuca, serce - 70-80%. Najmniej - 40% wody - zawarte jest w szkielecie. Makroelementy - węgiel, wodór, tlen, azot, siarka, fosfor - wchodzą w skład białek, kwasów nukleinowych i innych biologicznie aktywnych związków organizmu. Zawartość węgla w białkach wynosi 51-55%, tlenu - 22-24%, azotu - 15-18%, wodoru - 6,5-7%, siarki - 0,3-2,5%, fosforu - około 0,5%. Węgiel, wodór i tlen są również zawarte w składzie węglowodanów, których zawartość w tkankach zwierzęcych jest niewielka - około 2%. Pierwiastki te wchodzą w skład lipidów (tłuszczów). Ponadto fosfolipidy zawierają fosfor w postaci grup fosforanowych. Lipidy w największym stopniu koncentrują się w mózgu (12%), następnie w wątrobie (5%), mleku (2-3%) i surowicy krwi (0,6%). Jednak większość fosforu (600 g) zawarta jest w tkance kostnej. Stanowi to 85% masy całego fosforu występującego w organizmie człowieka. Fosfor koncentruje się także w twardych tkankach zębów, w których występuje wraz z wapniem, chlorem, fluorem w postaci hydroksylowej, chloru, fluoroapatytów o wzorze ogólnym Ca5 (PO4)3X, gdzie X = odpowiednio OH, CI, F. Wapń koncentruje się głównie w kościach, a także w tkankach zębów. Sód i chlor występują głównie w płynach pozakomórkowych, natomiast potas i magnez występują głównie w płynach wewnątrzkomórkowych. W postaci fluorków sód i potas wchodzą w skład tkanek kostnych i zębów. Magnez jako fosforan Mg3 (PO4)2 zawarte w twardych tkankach zęba. Hormony biorą udział w utrzymaniu w organizmie określonej zawartości makro- i mikroelementów. 34. Biologiczna rola pierwiastków chemicznych w organizmie Biologiczna rola pierwiastków chemicznych w organizmie człowieka jest niezwykle zróżnicowana. Główną funkcją makroskładników jest budowanie tkanek, utrzymywanie stałego ciśnienia osmotycznego, składu jonowego i kwasowo-zasadowego. Mikroelementy wchodzące w skład enzymów, hormonów, witamin, substancji biologicznie czynnych jako czynniki kompleksujące lub aktywatory biorą udział w metabolizmie, procesach rozmnażania, oddychaniu tkanek i neutralizacji substancji toksycznych. Mikroelementy aktywnie wpływają na procesy hematopoezy, utleniania, redukcji, przepuszczalności naczyń krwionośnych i tkanek. Makro i mikroelementy – wapń, fosfor, fluor, jod, glin, krzem – warunkują powstawanie tkanki kostnej i zębowej. Zidentyfikowano wiele chorób związanych z niedoborem lub nadmiernym nagromadzeniem różnych mikroelementów. Niedobór fluoru powoduje próchnicę zębów, niedobór jodu powoduje wole endemiczne, a nadmiar molibdenu powoduje endemiczną dnę moczanową. Tego rodzaju wzorce związane są z faktem, że organizm człowieka utrzymuje równowagę optymalnych stężeń pierwiastków biogennych – homeostazę chemiczną. Zaburzenie tej równowagi na skutek niedoboru lub nadmiaru tego pierwiastka może prowadzić do różnych chorób. Oprócz sześciu głównych makroelementów - organogenów (węgla, wodoru, azotu, tlenu, siarki i fosforu), które tworzą węglowodany, tłuszcze, białka i kwasy nukleinowe, makroelementy nieorganiczne są niezbędne do normalnego odżywiania ludzi i zwierząt - wapń, chlor , magnez, potas, sód - oraz pierwiastki śladowe - miedź, fluor, jod, żelazo, molibden, cynk, a także ewentualnie (sprawdzone na zwierzętach) selen, arsen, chrom, nikiel, krzem, cyna, wanad. Analiza zawartości i proporcji mikroelementów w organizmie człowieka wykorzystywana jest również w kryminalistycznych badaniach lekarskich. Na przykład w przypadku zatrucia alkoholem, pod wpływem alkoholu etylowego wzrasta zawartość wapnia w wątrobie, a sód i potas maleją. W tym samym czasie w sercu i nerkach zmniejsza się zawartość wapnia. Brak w diecie pierwiastków takich jak żelazo, miedź, fluor, cynk, jod, wapń, fosfor, magnez i niektórych innych prowadzi do poważnych konsekwencji dla zdrowia człowieka. Trzeba jednak pamiętać, że nie tylko niedobór, ale i nadmiar pierwiastków biogennych jest szkodliwy dla organizmu, gdyż zaburza to homeostazę chemiczną. Składniki mineralne, które są niezbędne w znikomych ilościach, stają się toksyczne w wyższych stężeniach. Wiele pierwiastków (srebro, rtęć, ołów, kadm itp.) Uważa się za toksyczne, ponieważ ich przedostanie się do organizmu, nawet w śladowych ilościach, prowadzi do poważnych zjawisk patologicznych. Różne pierwiastki i ich związki są szeroko stosowane jako leki. Tak więc badanie biologicznej roli pierwiastków chemicznych, wyjaśnienie związku między wymianą tych pierwiastków a innymi substancjami biologicznie czynnymi (enzymami, hormonami, witaminami) przyczynia się do tworzenia nowych leków i opracowania optymalnych schematów dawkowania dla zarówno w celach terapeutycznych, jak i profilaktycznych. 35. Pierwiastki S i ich związki Woda jest jednym z najważniejszych i najbardziej rozpowszechnionych związków wodoru na Ziemi. Przestrzeń wodna zajmuje prawie 75% powierzchni globu. Ciało osoby dorosłej zawiera średnio 65-67% wody, płodu (4 miesiące) - 94%, a noworodka - 74%. Wszystkie reakcje chemiczne w organizmie zachodzą tylko w środowisku wodnym. Życie bez wody jest niemożliwe. Woda destylowana jest preparatem farmakopealnym. W praktyce medycznej stosuje się inny związek wodoru - nadtlenek wodoru H2 02. Związek ten jest ważnym produktem ubocznym metabolizmu. Nadtlenek wodoru jest bezbarwną, przezroczystą cieczą. Powoduje pieczenie w kontakcie ze skórą i błonami śluzowymi. Cząsteczka H2О2 polarny. Obecność wolnych par elektronów na atomach tlenu umożliwia tworzenie wiązań donor-akceptor nadtlenku wodoru z ligandami - akceptorami elektronów. Stan utlenienia tlenu w H2О2 jest równy 1, tj. ma wartość pośrednią między stopniem utlenienia tlenu w wodzie (2) a tlenem pierwiastkowym O2 . Wynika z tego, że nadtlenek wodoru może wykazywać zarówno właściwości środka utleniającego, jak i właściwości środka redukującego (dwoistość redoks). Jednak sądząc po standardowych potencjałach połówkowej reakcji redoks, właściwości utleniające są bardziej charakterystyczne dla nadtlenku wodoru. Czysty nadtlenek wodoru jest termodynamicznie niestabilny i po postoju rozkłada się wybuchowo na wodę i tlen, uwalniając dużą ilość ciepła. Wodne roztwory nadtlenku wodoru są bardziej stabilne, w chłodnym miejscu można je przechowywać przez dłuższy czas. Nadtlenek wodoru sprzedawany jest najczęściej w postaci 30% roztworu wodnego – perhydrolu. Współproces rozkładu nadtlenku wodoru ulega znacznemu przyspieszeniu w obecności soli metali ciężkich. Katalizowany jonami metali rozkład nadtlenku wodoru może prowadzić do powstania rodników, z których najważniejszymi są wodorotlenek HO i wodoronadtlenek. Toksyczność związana jest z faktem, że H2О2 i O.2 oddziałują z warstwą lipidową błon komórkowych i uszkadzają je. W praktyce medycznej nadtlenek wodoru stosuje się głównie jako zewnętrzny środek bakteriobójczy. Akcja H2О2 opiera się na zdolnościach utleniających nadtlenku wodoru i nieszkodliwości produktu jego redukcji – wody. Podczas leczenia ran uwolniony tlen odgrywa podwójną rolę: 1) ma działanie przeciwdrobnoustrojowe, dezodoryzujące i depigmentujące, zabijając ciała drobnoustrojów; 2) tworzy pianę, przyczyniając się do przejścia cząstek rozpadu tkanek do stanu zawieszonego i oczyszczenia ran. Jako preparat farmakopealny stosuje się 3% wodny roztwór nadtlenku wodoru, do rozjaśniania włosów stosuje się 6% roztwór nadtlenku wodoru. W postaci 30% roztworu nadtlenek wodoru stosowany jest w leczeniu brodawkowatej postaci liszaja płaskiego oraz do usuwania młodzieńczych brodawek. 36. Biologiczna rola pierwiastków s z grupy IA (lit, rubid, cez, frans) Pod względem zawartości w organizmie człowieka sód (0,08%) i potas (0,23%) to makroelementy, a pozostałe metale alkaliczne to lit (104%), rubid (105%), cez (104%) - do pierwiastków śladowych. Lit Zawartość litu w organizmie człowieka wynosi około 70 mg (10 mmol) – 104%. Związki litu u zwierząt wyższych koncentrują się w wątrobie, nerkach, śledzionie, płucach, krwi i mleku. Maksymalna ilość litu znajduje się w ludzkich mięśniach. Biologiczna rola litu jako pierwiastka śladowego nie została jeszcze w pełni wyjaśniona. Udowodniono, że na poziomie błon komórkowych jony Li (w wystarczającym stężeniu) konkurują z jonami sodu podczas wnikania do komórek. Oczywiście zastąpienie jonów Na w komórkach jonami Li wiąże się z większą kowalencją związków litu, dzięki czemu lepiej rozpuszczają się w fosfolipidach. Ustalono, że niektóre związki litu mają pozytywny wpływ na pacjentów z depresją maniakalną. Wchłaniane z przewodu pokarmowego jony Li gromadzą się we krwi. Gdy stężenie jonów Li osiąga 0,6 mmol/l i więcej, następuje spadek napięcia emocjonalnego i osłabienie podniecenia maniakalnego. Jednocześnie zawartość jonów Li w osoczu krwi musi być ściśle kontrolowana. W przypadkach, gdy stężenie jonów Li przekracza 1,6 mmol/l, możliwe są zjawiska negatywne. Rubid i cez Według zawartości rubidu w ludzkim ciele (105%) i cezu (104%) należą do pierwiastków śladowych. Są stale zawarte w ciele, ale ich rola biologiczna nie została jeszcze wyjaśniona. Jako kompletny analog potasu, rubid gromadzi się również w płynie wewnątrzkomórkowym i może zastąpić równoważną ilość potasu w różnych procesach. izotopy radioaktywne 13rCS i 87Rb jest stosowany w radioterapii nowotworów złośliwych, a także w badaniu metabolizmu potasu. Dzięki szybkiemu rozpadowi można je nawet wprowadzić do organizmu bez obaw o długotrwałe szkodliwe działanie. Francja Jest to radioaktywny pierwiastek chemiczny otrzymywany sztucznie. Istnieją dowody na to, że frans może selektywnie gromadzić się w nowotworach na najwcześniejszych etapach ich rozwoju. Obserwacje te mogą być przydatne w diagnostyce raka. Zatem z pierwiastków grupy IA fizjologicznie aktywne są Li, Rb, Cs, a Na i K są niezbędne. Podobieństwo właściwości fizykochemicznych Li i Na, wynikające z podobieństwa budowy elektronowej ich atomów, objawia się także w biologicznym działaniu kationów (akumulacja w płynie zewnątrzkomórkowym, zamienność). Podobny charakter biologicznego działania kationów pierwiastków o długich okresach – K+, Rb+, CS+ (akumulacja w płynie wewnątrzkomórkowym, wymienność) wynika również z podobieństwa ich budowy elektronowej i właściwości fizykochemicznych. Stanowi to podstawę do stosowania preparatów sodowych i potasowych w przypadku zatrucia solami litu i rubidu. 37. Biologiczna rola pierwiastków s z grupy IA (sód, potas) Zawartość sodu w organizmie człowieka o masie 70 kg wynosi około 60 g (2610 mmol) - 0,08%. Z tej ilości 44% sodu znajduje się w płynie zewnątrzkomórkowym, a 9% w płynie wewnątrzkomórkowym. Pozostała ilość sodu znajduje się w tkance kostnej, która jest miejscem odkładania się jonów Na w organizmie. Około 40% sodu zawartego w tkance kostnej bierze udział w procesach metabolicznych, dzięki czemu szkielet jest dawcą lub akceptorem jonów Na, co pozwala na utrzymanie stałego stężenia jonów Na w płynie zewnątrzkomórkowym. Sód jest głównym jonem zewnątrzkomórkowym. W organizmie człowieka sód występuje w postaci rozpuszczalnych soli, głównie chlorków, fosforanów i wodorowęglanów. Sód rozprowadzany jest po całym organizmie: w surowicy krwi, płynie mózgowo-rdzeniowym, płynie ocznym, sokach trawiennych, żółci, nerkach, skórze, tkance kostnej, płucach, mózgu. Jony Na odgrywają ważną rolę w zapewnieniu stałości środowiska wewnętrznego organizmu człowieka, biorą udział w utrzymywaniu stałego ciśnienia osmotycznego biopłynu (homeostaza osmotyczna). Jony Na biorą udział w regulacji metabolizmu wody i wpływają na funkcjonowanie enzymów. Wraz z jonami K, Mg, Ca, Cl, jon Na bierze udział w przekazywaniu impulsów nerwowych i utrzymuje prawidłową pobudliwość komórek mięśniowych. Gdy zmienia się zawartość sodu w organizmie, dochodzi do dysfunkcji układu nerwowego, sercowo-naczyniowego i innych, mięśni gładkich i szkieletowych. Chlorek sodu NaCl jest głównym źródłem kwasu solnego do soku żołądkowego. Sód dostaje się do organizmu człowieka głównie w postaci soli kuchennej. Rzeczywiste dzienne zapotrzebowanie organizmu na sód wynosi 1 g, chociaż średnie spożycie tego pierwiastka sięga 4-7 g. Ciągłe nadmierne spożycie NaCl przyczynia się do nadciśnienia. Kiedy komórki drobnoustrojów są narażone na działanie zasad, następuje wytrącanie białek komórkowych, a w rezultacie śmierć mikroorganizmów. Siarczan sodu (sól glaubera) Na2SO4 × 10H2O jest używany jako środek przeczyszczający. Tetraboran sodu Na2B4О7 × 10H2O jest stosowany zewnętrznie jako środek antyseptyczny do płukania, podmywania, smarowania. Wodorotlenek sodu w postaci 10% roztworu jest częścią silinu stosowanego w praktyce ortopedycznej do odlewania modeli ogniotrwałych do produkcji protez odlewanych ze stopu kobaltowo-chromowego. Zawartość potasu w organizmie człowieka o masie 70 kg wynosi około 160 g (4090 mmol) - 0,23%. Potas jest głównym kationem wewnątrzkomórkowym, stanowiącym 2/3 wszystkich aktywnych kationów komórkowych. Z całkowitej ilości potasu zawartego w organizmie 98% znajduje się wewnątrz komórek, a tylko około 2% w płynie zewnątrzkomórkowym. Potas rozprowadzany jest po całym organizmie. Jego topografia: wątroba, nerki, serce, tkanka kostna, mięśnie, krew, mózg itp. Jony K odgrywają ważną rolę w procesach fizjologicznych - skurczu mięśni, prawidłowym funkcjonowaniu serca, przewodzeniu impulsów nerwowych, reakcjach metabolicznych. Jony K są ważnymi aktywatorami enzymów znajdujących się wewnątrz komórki. 38. Biologiczna rola pierwiastków s grupy IIA. Ich zastosowanie w medycynie (beryl, magnez, wapń) Beryl występuje zarówno w roślinach, jak iw organizmach zwierzęcych. Zawartość berylu w organizmach żywych wynosi 107%, czyli jest ultramikroelementem zanieczyszczenia. Biologiczna rola berylu nie została wystarczająco zbadana. Związki berylu są toksyczne i powodują szereg chorób (krzywica berylu, beryloza itp.). Szczególnie toksyczne są związki lotne berylu. Negatywny wpływ Be2 + na procesy fizjologiczne tłumaczy się jego właściwościami chemicznymi. Magnez jest formalnie klasyfikowany jako makroelement. Jego całkowita zawartość w organizmie wynosi 0,027% (około 20 g). Topografia magnezu w organizmie człowieka jest następująca: magnez koncentruje się w największym stopniu w zębinie i szkliwie zębów oraz tkance kostnej. Gromadzi się także w trzustce, mięśniach szkieletowych, nerkach, mózgu, wątrobie i sercu. U osoby dorosłej dzienne zapotrzebowanie na magnez wynosi około 0,7 g. Jon Mg, podobnie jak jon K, jest kationem wewnątrzkomórkowym. W płynach biologicznych i tkankach organizmu magnez występuje zarówno w postaci jonów wodnych, jak i w stanie związanym z białkiem w ilości < 102%, czyli w istocie jest mikroelementem. Stężenie jonów Mg wewnątrz komórek jest około 2,5-3 razy wyższe niż w płynach pozakomórkowych. Jony magnezu odgrywają ważną rolę biologiczną w organizmie człowieka. Ze względu na mniejszy promień jonów i wyższą energię jonizacji Mg2+ tworzy silniejsze wiązania niż jon Ca i dlatego jest bardziej aktywnym katalizatorem procesów enzymatycznych. Będąc częścią różnych układów enzymatycznych, jon Mg jest ich zasadniczym składnikiem i aktywatorem (enzymy takie jak karboksypeptydaza, cholinoesteraza i inne są specyficzne dla jonu Mg). Hydroliza ATP, związana z szeregiem reakcji enzymatycznych, w wyniku których powstaje hydrofosfation HPO2 i uwalniana jest duża ilość energii, zachodzi przy nadmiarze Mg2+. Wapń jest makroelementem. Jego całkowita zawartość w organizmie wynosi 1,4%. Wapń znajduje się w każdej komórce ludzkiego ciała. Większość wapnia znajduje się w kościach i tkankach zębów. Osoba dorosła powinna spożywać średnio 1 g wapnia dziennie, choć zapotrzebowanie na wapń to zaledwie 0,5 g. Wapń podawany z pokarmem wchłania się w jelitach tylko w 50%. Stosunkowo słabe wchłanianie jest konsekwencją powstawania w przewodzie pokarmowym trudno rozpuszczalnego fosforanu wapnia Ca3(PO4)2 oraz sole wapniowe kwasów tłuszczowych. W organizmie stężenie jonów Ca jest regulowane przez hormony. W kościach i zębach osoby dorosłej około 1 kg wapnia występuje w postaci nierozpuszczalnego krystalicznego minerału – hydroksyapatytu Ca10(RO4)6(OH)2 , którego powstawanie następuje podczas oddziaływania jonów Ca z fosforanami. We krwi i limfie wapń występuje zarówno w stanie zjonizowanym, jak i niezjonizowanym – w połączeniu z białkami, węglowodanami itp. Mechanizm krzepnięcia krwi składa się z kilku etapów, zależnych od obecności zjonizowanego Ca. Jony Ca biorą udział w przekazywaniu impulsów nerwowych, skurczu mięśni i regulacji pracy mięśnia sercowego. Stężenie jonów Ca wewnątrz i na zewnątrz komórki wynosi odpowiednio 106 oraz (2,25-2,8) 103 mol/l. Ponieważ wapń praktycznie nie jest wykorzystywany wewnątrz komórki, pełni rolę budulca w organizmie - w kościach i zębach. Szkielet jest głównym magazynem wapnia w organizmie. 39. Biologiczna rola pierwiastków d grupy VIB. Ich zastosowanie w medycynie Chrom znajduje się w organizmach roślinnych i zwierzęcych. Ciało dorosłego człowieka zawiera około 6 g Cr (0,1%). Metaliczny chrom jest nietoksyczny, natomiast związki Cr(III) i Cr(VI) są niebezpieczne dla zdrowia. Powodują podrażnienie skóry, co prowadzi do zapalenia skóry. Zakłada się, że pochodne chromu (VI) mają właściwości rakotwórcze. 0,25-0,3 g dwuchromianu potasu powoduje śmierć. Związki chromu (VI) stosuje się jako środki grzybobójcze (trawiacze, grzyby - „grzybowe”, caldere - „do zabijania”). Związki chromu(III) korzystnie wpływają na wzrost roślin. Molibden należy do „metali życia”, będąc jednym z najważniejszych biopierwiastków. Jej szczególną pozycję zauważyli 20-25 lat temu F. Krin i L. Oril. Naukowcy ci wysunęli ideę, że pojawienie się życia na Ziemi nie nastąpiło przez ewolucję, ale że zostało ono sprowadzone przez nieznaną cywilizację z kosmosu z gwiazd molibdenowych, gdzie życie istniało na długo przed nami. W procesach biochemicznych molibden bierze udział w stanach utlenienia VI i VI. W tych stanach tworzy stabilne formy okso. Molibden tworzy stabilne kompleksy okso i najwyraźniej dlatego jest częścią enzymów zapewniających przenoszenie grup okso. Mo (VI) dominuje we krwi; jeśli ligandem jest tlen, powstają stabilne izopolimolibdacje. Nadmierna zawartość molibdenu w żywności zaburza metabolizm Ca2+ i RO4 , powodując spadek wytrzymałości kości - osteoporoza. Prawdopodobnie dochodzi do wiązania się z kompleksami fosfomolibdenu. Takie kompleksy można uważać za reszty kwasowe kwasów heteropolimolibdenowych. W przypadku wapnia pozostałości te tworzą nierozpuszczalne kryształy. Jest możliwe, że te kryształy inicjują odkładanie się soli kwasu moczowego i powodują dnę moczanową. Dna deformuje stawy, co uzasadnia jej dosłowne tłumaczenie - „pułapka na stopy”. Oprócz kompleksów tlenowych molibden tworzy kompleksy halogenkowe (Hal), tiocyjanianowe (NCS) i cyjankowe (CN). Molibden jest składnikiem różnych enzymów. W ludzkim ciele są to hydroksydazy aldehydowe, dehydrogenazy ksantynowe i oksydazy ksantynowe. Masa cząsteczkowa oksydazy ksantynowej (COX) wynosi 250 000 a.u. np. Jest to enzym ssaków zawierający molibden. Może katalizować utlenianie ksantyny i innych puryn, a także aldehydów. Konwersja hipoksantyny i ksantyny do kwasu moczowego jest katalizowana przez oksydazę ksantynową. Przyjmuje się, że w procesie katalitycznym molibden tworzy wiązanie z azotem i tlenem ksantyny. Molibden jest najważniejszym mikroelementem roślin, ponieważ substancje biologicznie czynne z jego udziałem zapewniają łagodne wiązanie azotu: przekształcają go w amoniak lub produkty zawierające azot. W porównaniu z innymi metalami o znaczeniu przemysłowym molibden ma niską toksyczność. Spożycie molibdenu z pokarmem wynosi 0,1 - 0,3 mg / dzień, ale wymagane dzienne spożycie nie zostało ustalone. Niedobór molibdenu powoduje zmniejszenie aktywności oksydazy ksantynowej w tkankach. Nadmierna zawartość molibdenu powoduje osteoporozę. Wolfram jest pierwiastkiem śladowym. Jego rola w organizmie nie jest dobrze poznana. Anionowa forma wolframu jest łatwo wchłaniana w przewodzie pokarmowym. Metalowy wolfram i jego kationowe formy nie są wchłaniane przez organizm. Brak informacji na temat homeostazy wolframu u ssaków. 40. Biologiczna rola związków manganu. Ich zastosowanie w medycynie Spośród pierwiastków grupy VIIB tylko mangan jest pierwiastkiem biogennym i jednym z dziesięciu „metali życia” niezbędnych do normalnego przebiegu procesów w organizmach żywych. Ciało osoby dorosłej zawiera 12 mg. Mangan koncentruje się w kościach (43%), reszta w tkankach miękkich, w tym w mózgu. W organizmie mangan tworzy kompleksy metali z białkami, kwasami nukleinowymi, ATP, ADP, poszczególnymi aminokwasami. Zawierają metaloenzymy manganu arginazę, cholinoesterazę, fosfoglukomutazę, karboksylazę pirogronianową. Wiązanie amoniaku, toksycznego produktu przemian aminokwasów w organizmie ssaków, odbywa się poprzez aminokwas argininę. Arginaza jest enzymem katalizującym hydrolizę argininy w wątrobie. W rezultacie arginina rozkłada się na mocznik i cykliczny aminokwas ornitynę. Mocznik jest nietoksyczną, rozpuszczalną w wodzie substancją. Jest przenoszony z krwią do nerek i wydalany z moczem. Promień atomowy manganu wynosi 128 pm. Tłumaczy to fakt, że mangan może zastąpić magnez (promień atomowy 160 pm) w połączeniu z ATP, znacząco wpływając na transfer energii w organizmie. Jony Mg i Mn aktywują także enzymy - nukleazy. Enzymy te katalizują hydrolizę kwasów nukleinowych DNA i RNA w dwunastnicy. W efekcie te biopolimery rozszczepiają się na jednostki monomeru – nukleotydy. W szczególności taką nukleazą jest deoksyrybonukleaza, która katalizuje hydrolizę DNA dopiero w obecności Mg2+ lub Mn2+. Mangan może być również częścią związków nieorganicznych w organizmie. Jest to na przykład słabo rozpuszczalny pirofosforan manganowo-magnezowy MnMgP2O7. Kryształy tej soli zlokalizowane są na wewnętrznej powierzchni błony pęcherzyka. Prawie taka sama wartość promienia atomowego manganu i żelaza wyjaśnia zdolność manganu do zastępowania żelaza w kompleksie porfiryny erytrocytów. Z tego samego powodu mangan może również zastąpić cynk w enzymach zależnych od cynku, zmieniając w ten sposób ich właściwości katalityczne. Nadmanganian potasu KMnO4 - najsłynniejszy związek manganu stosowany w medycynie. Użyj roztworów wodnych zawierających KMnO4 0,01-5%. Jako środek hemostatyczny stosuje się 5% roztwór. Roztwory nadmanganianu potasu mają właściwości antyseptyczne, które determinuje jego wysoka siła utleniająca. Spośród innych związków manganu należy zwrócić uwagę na siarczan manganu(II) i chlorek manganu(II), które są stosowane w leczeniu anemii. Brak danych na temat obecności technetu w organizmach żywych. Natomiast w diagnostyce radioizotopowej stosuje się związki technetu z bisfosfonianami. 41. Biologiczna rola związków żelaza. Hemoglobina Żelazo jest pierwiastkiem biogennym występującym w tkankach zwierząt i roślin. Całkowita masa żelaza w ciele osoby dorosłej wynosi około 5 g, co stanowi 0,007%. Żelazo metaliczne jest mało toksyczne, a związki Fe(II), Fe(III) i Fe(VI) w dużych ilościach są niebezpieczne dla zdrowia. Mioglobina, cytochromy, katalaza zapewniają oddychanie komórkowe. Wszystkie te białka składają się z rzeczywistych części białkowych i związanych z nimi ośrodków aktywnych. Centrum aktywne stanowi makrocykliczny związek złożony – hem. Związek porfiryna działa jako ligand makrocykliczny. Atomy azotu dawcy znajdują się w rogach kwadratu, w środku którego znajduje się jon Fe. Ogólnie kompleks ma konfigurację oktaedryczną. Piąty orbital przechodzący przez azot aminokwasu (histydyna) służy do połączenia hemu z białkiem. Hemoglobina składa się z 4 cząsteczek białka (podjednostek), które tworzą pojedynczy agregat makrocząsteczkowy. Każda podjednostka ma podobną strukturę do cząsteczki mioglobiny. Zatem hemoglobina może jednocześnie wiązać cztery cząsteczki O2 i mioglobina - 1. W tkankach znajduje się także kilka kompleksów białkowych zawierających żelazo niehemowe. Są to na przykład enzymy – oksydazy, a także białka magazynujące (depoty) i transportery żelaza. Nadmiar żelaza jest transportowany we krwi przez białkową transferynę i gromadzi się w postaci białka ferrytyny w różnych tkankach i narządach, zwłaszcza w wątrobie, śledzionie i szpiku kostnym. Ferrytyna składa się z 24 cząsteczek białka (podjednostek), które tworzą kulę o średnicy 12-14 nm. Każda podjednostka zawiera wnękę o średnicy 7 nm zawierającą do 4500 atomów żelaza. W ten sposób każdy agregat ferrytyny może przechowywać około 100 000 atomów żelaza, zapewniając liczne reakcje metaboliczne z udziałem tego pierwiastka. Opierając się na prawach równowagi chemicznej, nietrudno zrozumieć funkcjonowanie hemoglobiny jako nośnika tlenu z płuc do tkanek. Hemoglobina bez tlenu (deoksyhemoglobina) jest słabym kwasem, a jej wzór chemiczny można przedstawić jako HHb+. Dodaniu tlenu towarzyszy eliminacja protonu i powstaje oksyhemoglobina HbO.2 . W tym przypadku istnieje równowaga: HHb+ +O2 → Hb O2 + H+. Kiedy uboga w tlen krew żylna dostaje się do płuc, gdzie ciśnienie parcjalne tlenu jest wysokie (do 20 kPa), jej rozpuszczalność wzrasta zgodnie z prawem Henry'ego. Prowadzi to, zgodnie z zasadą Le Chateliera, do przesunięcia równowagi w prawo i powstania oksyhemoglobiny. Dodatkowe przesunięcie równowagi w prawo wynika z faktu, że w płucach wzrasta wartość pH (do 7,5). W rezultacie w płucach deoksyhemoglobina jest prawie całkowicie (do 97%) nasycona tlenem i przechodzi do oksyhemoglobiny. W naczyniach włosowatych penetrujących tkanki obwodowe ciśnienie parcjalne tlenu spada do 5 kPa, a wartość pH spada do 7,2. W rezultacie równowaga przesuwa się w lewo. We krwi płynącej z obwodu hemoglobina jest nasycona tlenem tylko w 65%. 42. Biologiczna rola związków żelaza. Tlenek węgla CO. Właściwości kompleksów metali białek zawierających hem ujawniają się pod wpływem substancji toksycznych, takich jak CO (tlenek węgla) i MCN (cyjanek – sole kwasu cyjanowodorowego). Najważniejsze z fizjologicznego punktu widzenia są białka zawierające żelazo: hemoglobina, mioglobina, cytochromy, peroksydazy, katalaza. Hemoglobina jest głównym składnikiem czerwonych krwinek, zapewnia oddychanie zewnętrzne, będąc nośnikiem tlenu z płuc do tkanek. Żelazo Fe i kobalt Co to niezbędne pierwiastki śladowe organizmów żywych. Tlenek węgla CO jest jednym z produktów niepełnego spalania paliwa. Znaczne ilości tego gazu wydzielają się podczas pracy kotłów, silników spalinowych i palenia. Gdy CO jest wdychany z powietrzem w płucach równolegle z oksyhemoglobiną HbO2 powstaje związek kompleksowy metalu - karbonylohemoglobina HbCO. Stała stabilności HbCO jest około 200 razy większa niż HbO2 . Dlatego nawet niewielkie ilości CO „przechwytują” znaczną część deoksyhemoglobiny, w efekcie czego zmniejsza się dopływ tlenu do narządów. Pojawiają się oznaki niedotlenienia – niedoboru tlenu. W pierwszej kolejności dotknięte są tkanki nerwowe. Aby odtruć (wyeliminować toksyczne działanie) tlenku węgla, w wielu przypadkach wystarczy przerwać jego dopływ i zwiększyć wentylację tlenową – wyprowadzić poszkodowanego na świeże powietrze. W tym przypadku zasada Le Chateliera znów się sprawdza – równowaga przesuwa się w stronę tworzenia oksyhemoglobiny. W wysokich stężeniach tlenek węgla blokuje białka oddychania komórkowego zawierające hem i trudno jest uniknąć śmierci. Mechanizm działania cyjanków jest podobny, ale ich toksyczność jest wyższa niż CO. Dostanie się do krwi nawet bardzo niewielkich ilości tych substancji prowadzi do zatrzymania oddechu i śmierci. Wysoką toksyczność cyjanków tłumaczy się dużą wytrzymałością wiązania Fe-CN, co warunkuje większą stabilność hemoglobiny cyjankowej. Oddychanie tlenu prowadzi do powstania nadtlenku wodoru H2O2 . Substancja ta ma wysoką zdolność utleniającą. Kiedy wchodzi w interakcję ze związkami bioorganicznymi komórek, powstają rodniki - bardzo aktywne cząstki molekularne o nienasyconej wartościowości i inicjowane jest utlenianie nadtlenku. Pod wpływem rodników niszczone są najważniejsze składniki komórki – błony i DNA. Podczas ewolucji biologicznej natura wykształciła specjalne białko – enzym katalazę, który niszczy nadtlenek wodoru. Ogranicza to nadmierne gromadzenie się tej substancji i zapobiega niszczeniu komórek. Działanie katalazy (CatFe2+ ) można przedstawić jako cykl katalityczny dwóch następujących po sobie reakcji: catfe2+ + H2O2 - Kot Fe2+ × H2O2 , catfe2+ × H2O2 + H2O2 → CatFe2+ + 2H2O2 +O2 . W rezultacie 2 cząsteczki nadtlenku wodoru zostają zniszczone, a cząsteczka biokatalizatora CatFe2+ zostaje uwolniona i może wejść w kolejny cykl katalityczny. Ten proces jest bardzo szybki. W ciągu sekundy 1 cząsteczka katalazy może wykonać do 20 000 cykli. 43. Biologiczna rola związków żelaza i kobaltu Przy braku żelaza w organizmie może rozwinąć się choroba - niedokrwistość z niedoboru żelaza (niedokrwistość). Istnieje niedobór tlenu w tkankach związany z brakiem żelaza do syntezy hemoglobiny. W rezultacie zmniejsza się dostarczanie tlenu do narządów obwodowych, a zatem zmniejsza się poziom oddychania komórkowego, a metabolizm zwalnia. Wprowadzenie chlorku żelaza (II) lub siarczanu żelaza (II) jako leków zmniejsza nasilenie choroby. W tym samym celu stosuje się drobny proszek metalicznego żelaza (żelazo zredukowane, do 1 g na dawkę), który jest łatwo rozpuszczalny w kwasie solnym soku żołądkowego. Dlatego działanie tego leku jest podobne do działania chlorku żelaza (II). Jednak leki będące bionieorganicznymi kompleksami żelaza z cukrami, nikotynamidem i innymi substancjami organicznymi są bardziej skuteczne. Takie kompleksy dobrze wchłaniają się do krwi, co jest przyczyną ich farmakologicznej skuteczności. Warto zauważyć, że od czasów starożytnych do czasów współczesnych w leczeniu anemii z niedoboru żelaza stosowano tzw. wino żelazne, czyli napój sporządzany poprzez zaparzanie wina gronowego na opiłkach żelaza. Oczywiście żelazo rozpuszcza się w winie (środku kwaśnym) i tworzy kompleksy z naturalnymi substancjami organicznymi, których zawiera w dużych ilościach. Oczywiste jest, że mechanizm działania starożytnego napoju jest w przybliżeniu taki sam jak współczesnych leków. Podobnie jak żelazo, kobalt jest również jednym z najważniejszych pierwiastków biogennych. Całkowita masa kobaltu w ciele osoby dorosłej wynosi około 1,2 mg, czyli mniej niż 10%. Około 100 mg tej masy występuje w postaci cyjanokobalaminy (witaminy B rozpuszczalnej w tłuszczach)12 ) i jego analogi. Substancja ta, podobnie jak hem, jest makrocyklicznym związkiem złożonym. Związek tetrakleszczowy, porfina, działa jako ligand makrocykliczny. R jest złożonym podstawnikiem organicznym. W analogach cyjanokobalaminy zamiast anionu CN działają różne podstawniki organiczne. Najważniejsza rola witaminy B12 odgrywa rolę w rozwoju i tworzeniu czerwonych krwinek (erytropoeza). Niedobór witaminy B12 (spożycie mniej niż 3 mcg dziennie) prowadzi do poważnej choroby - anemii złośliwej (niedokrwistość). Ustalono, że analogi cyjanokobalaminy są aktywatorami - kofaktorami różnych enzymów biorących udział w erytropoezie. Brak kofaktorów objawia się niedoborem hemoglobiny i erytrocytów. Rośliny i zwierzęta nie potrafią syntetyzować witaminy B12. Jest produkowany tylko przez niektóre rodzaje bakterii. Bakterie te są obecne w przewodzie pokarmowym człowieka. Syntetyzują wystarczającą ilość witaminy B12. Niedokrwistość złośliwa wiąże się z upośledzeniem wchłaniania tej witaminy do krwi. Dlatego przyjmowanie tabletek jest nieskuteczne. Wstrzyknięcie witaminy (100-200 mcg przez 2 dni) do krwi znacznie poprawia stan pacjenta z niedokrwistością złośliwą. 44. Rola elementów d grupy IB. Wykorzystanie ich związków w medycynie Miedź Cu jest mikroelementem niezbędnym dla organizmów żywych. Srebro Ag i złoto Au są pierwiastkami śladowymi. Ich związki znajdują zastosowanie w medycynie. Miedź jest pierwiastkiem biogennym występującym w tkankach zwierząt i roślin. Całkowita masa miedzi w organizmie dorosłego człowieka wynosi około 100 mg, co stanowi około 0,0001%. Około 30% tej ilości znajduje się w mięśniach. Wątroba i mózg są również bogate w miedź. Miedź metaliczna i jej związki są toksyczne. Do najważniejszych z fizjologicznego punktu widzenia należą białka zawierające miedź – oksydaza cytochromowa i dysmutaza ponadtlenkowa. Oksydaza cytochromowa jest jednym ze składników łańcucha oddechowego zlokalizowanego w błonach mitochondrialnych. Zapewnia oddychanie komórkowe poprzez redukcję tlenu do wody na końcu łańcucha oddechowego. Organizm potrzebuje 2,5-5,0 mg miedzi dziennie. Jeśli w organizmie brakuje miedzi, może rozwinąć się choroba - niedokrwistość z niedoboru miedzi. Miedź jest niezbędna do wchłaniania żelaza, zwłaszcza w syntezie oksydazy cytochromowej, która zawiera zarówno żelazo, jak i miedź. W przypadku niedoboru miedzi normalny rozwój tkanki łącznej i naczyń krwionośnych zostaje zakłócony. Zatrucie zwykle wiąże się z przypadkowym przedawkowaniem środków owadobójczych, wdychaniem proszku metalu, spożyciem roztworów soli miedzi. Duże niebezpieczeństwo stanowią napoje przechowywane w miedzianych naczyniach bez ochronnej powłoki ścian. Jako czynnik zewnętrzny stosuje się 0,25% wodny roztwór siarczanu miedzi CuSO.4 z zapaleniem błon śluzowych i zapaleniem spojówek. Małe dawki tego leku można przyjmować z jedzeniem, aby zwiększyć erytropoezę w anemii. Srebro i złoto W ciele osoby dorosłej znajduje się około 1 mg srebra, czyli około 10% (1 ppm) i do 10 mg złota, czyli około 10% (10 ppm). Właściwości antyseptyczne rozpuszczalnych soli srebra znane są od czasów starożytnych. Kapłani od dawna wiedzą, że woda ("święta") przechowywana w srebrnych naczyniach nie psuje się przez długi czas, to znaczy nie ulega skażeniu mikrobiologicznemu. Obecnie ta właściwość „srebrnej” wody jest wykorzystywana przez żeglarzy podczas dalekich rejsów. Silne objawy toksyczne u osoby dorosłej obserwuje się po spożyciu 7 g AgNO.3. W medycynie od dawna stosuje się leki takie jak krystaliczny azotan srebra AgNO03 (lapis) i jego wodne roztwory. Od dawna znane są preparaty koloidalnego srebra metalicznego protargol (8% Ag) i collargol (70% Ag), które są drobnymi proszkami o metalicznym połysku. Każda cząsteczka takich proszków jest kryształem zredukowanego metalicznego srebra o wielkości poniżej 1 μm z powłoką białkową z albuminy (protargol) lub kolagenu (kolargol). Otoczka białkowa chroni kryształki srebra przed sklejaniem się i zapewnia ich przejście do środowiska wodnego (solubilizuje). Preparaty ze srebra stosuje się jako środki przeciwzapalne, antyseptyczne i ściągające. Preparaty ze złota są również stosowane jako skuteczne leki przeciwzapalne. Najbardziej znane to krizanol z 30% zawartością metalu szlachetnego oraz złoto koloidalne. 45. Biologiczna rola pierwiastków d z grupy IIB. Wykorzystanie ich związków w medycynie Cynk Zn, kadm Cd, rtęć Hg to pierwiastki śladowe. Ciało dorosłego człowieka zawiera 1,8 g Zn, 50 mg Cd, 13 mg Hg. Kadm i rtęć są pierwiastkami zanieczyszczającymi. Około 70% rtęci koncentruje się w tkance tłuszczowej i mięśniowej. Kadm zlokalizowany jest w 30% w nerkach, pozostała część w wątrobie, płucach i trzustce. Cynk jest niezbędnym pierwiastkiem dla wszystkich roślin i zwierząt. W ciele osoby dorosłej większość cynku znajduje się w mięśniach (65%) i kościach (20%). Reszta ilości spada na osocze krwi, wątrobę, erytrocyty. Najwyższe stężenie cynku w gruczole krokowym. Cynk nie wykazuje zmiennej wartościowości. Najwyraźniej więc jego biokompleksy biorą udział w wielu reakcjach hydrolizy biochemicznej, które zachodzą bez przeniesienia elektronu. Jon Zn jest częścią ponad 40 metaloenzymów, które katalizują hydrolizę estrów i białek. Jednym z najbardziej przebadanych jest bionieorganiczny kompleks cynkowy – enzym anhydraza węglanowa (Mg = 30 000), składający się z około 260 reszt aminokwasowych. Cynk nie wchodzi w skład dipeptydaz – enzymów katalizujących hydrolizę dipeptydów (substancji składających się z 2 aminokwasów). Cynk tworzy bionieorganiczny kompleks z insuliną, hormonem regulującym poziom cukru we krwi. Zapotrzebowanie człowieka na cynk w pełni pokrywają produkty spożywcze: mięso, nabiał, jaja. Przy braku cynku w roślinach metabolizm białek i węglowodanów zostaje zakłócony, a synteza chlorofilu i witamin zostaje zahamowana. Niedobór cynku eliminowany jest poprzez stosowanie nawozów zawierających cynk. Toksyczność związków grupy IIB wzrasta od cynku do rtęci. Związki rozpuszczalne w wodzie działają drażniąco na skórę i powodują zatrucie w przypadku połknięcia. Same metale są również toksyczne - podczas wdychania par cynku (powietrza z produkcji cynku) pojawia się gorączka „metalowa”. Zatrucie oparami rtęci w średniowieczu nazywano „chorobą szalonego kapelusznika”. Poziom rtęci w żywności (owocach morza, np. w Japonii) prowadzi do choroby Minomata. Toksyczność rtęci jest związana z aglutynacją (sklejaniem się) czerwonych krwinek i hamowaniem enzymów. Przykładowo sublimacja powoduje zmianę wielkości, kruchość osmotyczną i zmniejszenie odkształcalności czerwonych krwinek, co jest niezbędne do ich przemieszczania się przez naczynia włosowate. Toksyczność kadmu jest związana z jego powinowactwem do kwasów nukleinowych. W wyniku przyłączenia się do DNA jego funkcjonowanie zostaje zakłócone. Przewlekła toksyczność kadmu i rtęci może zaburzać mineralizację kości. Pierwiastki toksyczne mogą zastąpić wapń. Prowadzi to do powstania apatytu o niedoskonałej strukturze na skutek zniekształcenia parametrów składnika krystalicznego tkanki kostnej. W rezultacie zmniejsza się wytrzymałość kości. 46. Właściwości toksyczne związków z grupy IIB (Zn, Cd, Hg) Związki Zn, Cd, Hg mogą powodować naruszenie metabolizmu białek, co objawia się uwalnianiem białek osocza przez nerki (w białkomoczu). Toksyczny wpływ związków z grupy IIB na organizm jest również spowodowany faktem, że te jony metali oddziałują z grupami sulfhydrylowymi SH białek, enzymów i aminokwasów. Kiedy jony metali oddziałują z grupami SH, powstają słabo dysocjujące i z reguły nierozpuszczalne związki. Dlatego blokowanie grup sulfhydrylowych prowadzi do tłumienia aktywności enzymów i zwijania białek. Jony metali dwuwartościowych blokują jednocześnie dwie grupy SH. W reakcjach tego typu jony metali pełnią rolę akceptora, a siarka pełni rolę donora elektronów. Najbardziej wyraźne powinowactwo chemiczne do grup SH w rtęci. Wynika to oczywiście z faktu, że rtęć ma wyższe właściwości kompleksujące i tworzy silniejsze wiązania z siarką. Grupy SH są częścią ponad 100 enzymów, których aktywność można stłumić poprzez blokowanie tych grup. Dlatego oczywiste jest, jak ważna jest znajomość mechanizmu blokowania i metod leczenia zatrucia organizmu metalami. Wiadomo, że toksyczne właściwości pierwiastków zależą od formy chemicznej, w jakiej dostają się do organizmu. Najbardziej toksyczne formy to te, które rozpuszczają się w lipidach i łatwo przenikają przez błonę do wnętrza komórki. W literaturze opisano przypadek masowego zatrucia rtęcią w Japonii. Nieorganiczne związki rtęci zostały przekształcone w metylortęć pod wpływem enzymów mikrobiologicznych. Metylortęć kumulowała się w rybach, a następnie wnikała do organizmu człowieka wraz z pożywieniem. Stopniowo koncentrując się, metylortęć powoduje nieodwracalne zniszczenie w ciele i śmierć. Zastosowanie związków cynku i rtęci w medycynie opiera się na ich działaniu ściągającym, kauteryzującym i antyseptycznym. Jako krople do oczu stosuje się 0,25% wodny roztwór siarczanu cynku ZnSO.4. W stomatologii chlorek cynku jest stosowany do przyżegania brodawczaków, w leczeniu stanów zapalnych błon śluzowych. Stosowany jest również tlenek cynku ZnO. Chlorek rtęci (II) (chlorek rtęci) jest bardzo toksyczny, a jego wodne roztwory w wysokich rozcieńczeniach (1:1000) służą do dezynfekcji. W leczeniu chorób skóry i chorób przenoszonych drogą płciową stosuje się maści zawierające tlenek rtęci (II) HgO i siarczek rtęci (II) HgS. Chlorek rtęci(I) (kalomel) jest słabo rozpuszczalny w wodzie i dlatego lekko toksyczny. Sól ta jest stosowana w weterynarii jako środek przeczyszczający. Rtęć w normalnych warunkach jest ciekłym metalem, który może rozpuszczać inne metale. W tym przypadku powstają twarde stopy - amalgamaty. W stomatologii do wypełniania zębów od dawna stosuje się amalgamaty srebra i kadmu. Są chemicznie obojętne, łatwo miękną po podgrzaniu i dlatego łatwo je kształtować. Źródła światła ultrafioletowego – lampy rtęciowo-kwarcowe do użytku medycznego – zawierają rtęć w postaci gazowej (pary). Naświetlanie światłem tych lamp w pomieszczeniach szpitalnych powoduje zniszczenie mikroorganizmów znajdujących się w powietrzu. Różne choroby skóry leczy się za pomocą promieni ultrafioletowych. Tak więc, w zależności od charakteru funkcjonowania i oddziaływania na organizm, metale z grupy IIB można podzielić na pierwiastek życiowy Zn oraz pierwiastki toksyczne Cd i Hg. 47. Biologiczna rola pierwiastków p z grupy IIIA. Wykorzystanie ich związków w medycynie Bor jest zanieczyszczeniem pierwiastkiem śladowym, jego udział masowy w ludzkim ciele wynosi 105%. Bor koncentruje się głównie w płucach (0,34 mg), tarczycy (0,30 mg), śledzionie (0,26 mg), wątrobie, mózgu (0,22 mg), nerkach, mięśniu sercowym (0,21 mg). Biologiczny wpływ boru nie został jeszcze dostatecznie zbadany. Wiadomo, że bor występuje w zębach i kościach, najwyraźniej w postaci trudno rozpuszczalnych soli kwasu borowego z kationami metali. Nadmiar boru jest szkodliwy dla organizmu człowieka. Istnieją dowody na to, że nadmiar boru hamuje amylazy, proteinazy i obniża aktywność adrenaliny. Według zawartości w ludzkim ciele (105%) aluminium należy do mikroelementów zanieczyszczeń. Aluminium koncentruje się głównie w surowicy krwi, płucach, wątrobie, kościach, nerkach, paznokciach, włosach, wchodzi w struktury osłonek nerwowych ludzkiego mózgu. Dzienne spożycie aluminium przez człowieka wynosi 47 mg. Aluminium wpływa na rozwój tkanek nabłonkowych i łącznych, regenerację tkanki kostnej, wpływa na wymianę fosforu. Aluminium ma wpływ na procesy enzymatyczne. Nadmiar glinu w organizmie hamuje syntezę hemoglobiny, ponieważ ze względu na dość wysoką zdolność kompleksowania aluminium blokuje centra aktywne enzymów biorących udział w hematopoezie. Istnieją dowody, że glin może katalizować reakcję transaminacji. Gal jest pierwiastkiem śladowym zanieczyszczenia (zawartość w organizmie człowieka wynosi 10-6-10-5%). Biologiczna rola galu w żywych organizmach jest prawie niejasna. Tal to wysoce toksyczny pierwiastek. Jon T1 ma tendencję, podobnie jak Ag+, do tworzenia silnych związków z ligandami zawierającymi siarkę. W efekcie jest bardzo toksyczny, gdyż hamuje aktywność enzymów zawierających grupy tiolowe – SH. Nawet bardzo małe ilości związków T1+ po spożyciu powodują wypadanie włosów. Ze względu na bliskość promieni K+ i T1+ mają podobne właściwości i są w stanie zastąpić się nawzajem w enzymach. Jony T1 i K to synergetyki. Tłumaczy to fakt, że enzymy kinaza pirogronianowa i dehydrataza diolowa są aktywowane nie tylko przez jony K, ale także jony T1 (jon T1 zastępuje jon K w centrum katalitycznym enzymów). Synergizm talu i potasu przejawia się również w tym, że podobnie jak jony K, jony T1 gromadzą się w erytrocytach. Jako antidotum na zatrucie jonami T1 stosuje się ligand zawierający siarkę, aminokwas cystynę. Podsumowując, należy zauważyć, że biologiczna rola pierwiastków p z grupy IIIA nie została dostatecznie zbadana. Obecnie wiadomo, że bor i gal oddziałują w roślinach z polifenolami, inhibitorami ich rozwoju, zmniejszając toksyczność tych ostatnich. Ustalono również niewątpliwą rolę glinu w budowie tkanek nabłonkowych i łącznych, a ponadto jego udział w procesach enzymatycznych, zarówno jako aktywator, jak i inhibitor. Jon T1 ma zdolność hamowania wielu enzymów zawierających siarkę. Aktywność biologiczna pierwiastków grupy IIIA związana jest głównie z ich zdolnością do tworzenia złożonych związków z ligandami zawierającymi tlen i nierozpuszczalnymi fosforanami. 48. Biologiczna rola pierwiastków p grupy IVA. Wykorzystanie ich związków w medycynie Zgodnie z zawartością w organizmie człowieka (21,15%) węgiel należy do makroelementów. Jest częścią wszystkich tkanek i komórek w postaci białek, tłuszczów, węglowodanów, witamin, hormonów. Z biologicznego punktu widzenia węgiel jest organogenem numer 1. Zgodnie z zawartością w ludzkim ciele (103% ־) krzem należy do mikroelementów zanieczyszczeń. Większość krzemu w wątrobie, nadnerczach, włosach, soczewkach. Ponieważ naturalny dwutlenek krzemu jest słabo rozpuszczalny w wodzie, przedostaje się do organizmu człowieka nie tyle przez przewód pokarmowy, ile drogą powietrzną przez płuca w postaci pylistego SiO2. Z naruszeniem metabolizmu krzemu wiąże się występowanie nadciśnienia, reumatyzmu, wrzodów, anemii. W praktyce medycznej stosuje się węglik krzemu (IV) SiC - karborund do szlifowania wypełnień i protez z tworzyw sztucznych. Dwutlenek krzemu SiO2 część cementów silikatowych. Należy zaznaczyć, że pył składający się z cząstek węgla, dwutlenku krzemu i glinu, systematycznie narażony na kontakt z płucami, powoduje chorobę – pylicę płuc. Narażenie na pył węglowy powoduje antrakozę, chorobę zawodową górników. W przypadku wdychania pyłu zawierającego S1O2 , pojawia się krzemica, a pod wpływem pyłu aluminium - glinoza. Według zawartości w ludzkim ciele (10-6-10-5%) german jest mikroelementem. Rola biologiczna nie została w pełni wyjaśniona. Związki germanu nasilają procesy hematopoezy w szpiku kostnym. Wiadomo również, że związki germanu mają niską toksyczność. Według zawartości w ludzkim ciele (104 %) cyna odnosi się do pierwiastków śladowych. Cyna wchodzi do organizmu człowieka wraz z kwaśną żywnością przechowywaną w blaszanych puszkach pokrytych warstwą cyny. W środowisku kwaśnym cyna rozpuszcza się i wchodzi do krwi w postaci soli, wykazując działanie toksyczne. Jednak w doświadczeniach na szczurach stwierdzono, że cyna w niewielkich ilościach ma stymulujący wpływ na wzrost szczurów. Daje to powód do przyjęcia jego konieczności dla ludzi. Niewątpliwie wyjaśnienie biologicznej roli tego mikroelementu wymaga dalszych badań. W praktyce medycznej stosuje się różne materiały, w szczególności materiały wypełniające zawierające cynę. Tak więc cyna jest częścią amalgamatu srebra (28%) do produkcji nadzień. Ołów i jego związki, zwłaszcza organiczne, są silnie toksyczne. Związki ołowiu wpływają na syntezę białek, bilans energetyczny komórki i jej aparat genetyczny. Wiele czynników sprzyja mechanizmowi denaturacji. Ustalono, że ołów jest jednym z pierwiastków, których obecność w pożywieniu wpływa na rozwój próchnicy. Z pożywieniem, wodą i powietrzem atmosferycznym człowiek wchłania dziennie do 100 mcg ołowiu. Ołów odkłada się głównie w szkielecie (do 90%) w postaci trudno rozpuszczalnego fosforanu. Udział masowy ołowiu w organizmie człowieka wynosi 106%. Za bezpieczne dla człowieka dzienne spożycie ołowiu wynosi 0,2–2 mg. W praktyce medycznej octan ołowiu (płyny) i tlenek ołowiu (II) PbO (część prostego plastra ołowianego) znalazły zastosowanie jako zewnętrzne, ściągające środki antyseptyczne. 49. Biologiczna rola pierwiastków p grupy VA. Wykorzystanie ich związków w medycynie (azot, fosfor) Zawartość azotu w organizmie człowieka (3,1%) należy do makroelementów. Jeśli weźmiemy pod uwagę tylko masę suchej masy organizmu (bez wody), wówczas zawartość azotu w komórkach wynosi 8-10%. Pierwiastek ten jest składnikiem aminokwasów, białek, witamin i hormonów. Azot tworzy wiązania polarne z atomami wodoru i węgla w biocząsteczkach. W wielu kompleksach bionieorganicznych (metaloenzymach) atomy azotu wiążą nieorganiczne i organiczne części cząsteczki poprzez mechanizm donor-akceptor. Razem z tlenem i węglem azot tworzy niezbędne związki - aminokwasy, które zawierają jednocześnie grupę aminową o właściwościach zasadowych i grupę karboksylową (-COOH) o właściwościach kwasowych. Grupa aminowa pełni także bardzo ważną funkcję w cząsteczkach kwasu nukleinowego. Fizjologiczne znaczenie bioligandów zawierających azot - porfiryn, na przykład hemoglobiny, jest ogromne. Cykl azotowy odbywa się w biosferze. Cykl azotu ma kluczowe znaczenie dla rolnictwa. Należy zwrócić uwagę na jeszcze jedną biologicznie ważną właściwość azotu - jego rozpuszczalność w wodzie jest prawie taka sama jak tlenu. Obecność nadmiaru azotu we krwi może być przyczyną rozwoju choroby dekompresyjnej. Wraz z gwałtownym wzrostem nurków następuje gwałtowny spadek ciśnienia, odpowiednio zmniejsza się rozpuszczalność azotu we krwi (prawo Henry'ego), a pęcherzyki azotu pierwiastkowego opuszczające krew zatykają małe naczynia, co może prowadzić do paraliżu i śmierci. Fosfor, ze względu na jego zawartość w organizmie człowieka (0,95%), zaliczany jest do makroskładników. Fosfor jest pierwiastkiem organogennym i odgrywa niezwykle ważną rolę w metabolizmie. W postaci fosforanu fosfor jest niezbędnym składnikiem wewnątrzkomórkowego ATP. Wchodzi w skład białek (0,5-0,6%), kwasów nukleinowych, nukleotydów i innych związków biologicznie czynnych. Fosfor jest podstawą szkieletu zwierząt i człowieka (ortofosforan wapnia, hydroksyapatyt), zębów (hydroksyapatyt, fluoroapatyt). Wiele reakcji biosyntezy zachodzi w wyniku przeniesienia grup fosforanowych z akceptora wysokoenergetycznego na niskoenergetyczny. System buforów fosforanowych jest jednym z głównych systemów buforowych we krwi. Żywe organizmy nie mogą obejść się bez fosforu. Znaczenie fosforu polega na tym, że cukry i kwasy tłuszczowe nie mogą być wykorzystywane przez komórki jako źródło energii bez uprzedniej fosforylacji. Wymiana fosforu w organizmie jest ściśle związana z wymianą wapnia. Potwierdza to zmniejszenie ilości nieorganicznego fosforu wraz ze wzrostem zawartości wapnia we krwi (antagonizm). Dzienne zapotrzebowanie człowieka na fosfor wynosi 1,3 g. Fosfor jest tak rozpowszechniony w produktach spożywczych, że przypadki jego oczywistego niedoboru (głodu fosforanowego) są praktycznie nieznane. Jednak nie cały fosfor zawarty w produktach spożywczych może zostać wchłonięty, gdyż jego wchłanianie zależy od wielu czynników: pH, stosunku zawartości wapnia do fosforu w żywności, obecności kwasów tłuszczowych w żywności, ale przede wszystkim od zawartości witaminy D. Jako leki stosuje się szereg związków fosforu. Należy zauważyć, że związki fosforoorganiczne zawierające wiązanie C-P są silnymi truciznami nerwowymi i są częścią bojowych środków chemicznych. 50. Biologiczna rola pierwiastków p grupy VA (arsen, antymon, bizmut). Ich zastosowanie w medycynie Zgodnie z zawartością w ludzkim ciele arszenik należy do pierwiastków śladowych. Koncentruje się w wątrobie, nerkach, śledzionie, płucach, kościach, włosach. Większość arsenu znajduje się w tkance mózgowej i mięśniach. Arsen gromadzi się w kościach i włosach i nie jest z nich całkowicie usuwany przez kilka lat. Ta cecha jest wykorzystywana w badaniach kryminalistycznych w celu wyjaśnienia kwestii, czy doszło do zatrucia związkami arsenu. Oznaczanie arsenu w materiale biologicznym przeprowadza się w prostym urządzeniu według reakcji Marsha: do obiektu biologicznego dodaje się cynk i kwas solny. Wodór uwolniony podczas reakcji redukuje dowolny związek arsenu do arsenu. Jeśli uwolniony wodór zawiera domieszkę arsenu, to AsH rozkłada się podczas ogrzewania mieszaniny gazowej3 : 2AsH3 = 2As° + 3H2. oraz czarna błyszcząca powłoka arsenu tworzy się na ściankach rurki uwalniającej gaz - „lustro arsenu”. Reakcja Marsha jest bardzo czuła i pozwala wykryć 7-107 g arsen. W stosunkowo dużych dawkach związki arsenu są bardzo toksyczne. Jak już wspomniano, toksyczne działanie związków arsenu wynika z blokowania grup sulfhydrylowych enzymów i innych substancji biologicznie czynnych. Ze względu na zawartość w organizmie człowieka (10%) antymon i bizmut zaliczane są do mikroelementów. Według klasyfikacji V.V. Kovalsky'ego antymon i bizmut zaliczane są do grupy mikroelementów stale występujących w organizmach żywych, ale których rola fizjologiczna i biochemiczna jest praktycznie nieznana. Fizjologiczna rola antymonu jest oczywiście podobna do arszeniku. Jony arsenu As i antymonu Sb oraz, w mniejszym stopniu, bizmutu Bi są synergetykami. Wiadomo zatem, że w prowincjach biogeochemicznych z nadmiarem arsenu w organizmach wzrasta zawartość nie tylko arsenu, ale także antymonu. Jednocześnie oba pierwiastki gromadzą się w tarczycy mieszkańców, hamują jej funkcję i powodują endemiczne wole. Synergizm arsenu i antymonu jest związany z ich zdolnością do tworzenia związków z ligandami zawierającymi siarkę. Bizmut jest bardziej skłonny do wiązania się z ligandami zawierającymi grupy aminowe. Tak więc wnikanie rozpuszczalnych związków bizmutu do organizmu prowadzi do zahamowania enzymów amino i karboksypolipeptydazy. Spożycie rozpuszczalnych w wodzie związków antymonu, takich jak stibina SbH3, ma działanie toksyczne podobne do związków arsenu. Związki bizmutu są również toksyczne po wstrzyknięciu. Na przykład dla psów dawka śmiertelna wynosi 6 mg/kg masy ciała. Jednak gdy większość związków antymonu i bizmutu dostanie się do przewodu pokarmowego, praktycznie nie mają działania toksycznego. Słaba toksyczność tych związków wynika z faktu, że sole Sb(III), Bi(III) w przewodzie pokarmowym ulegają hydrolizie, tworząc produkty słabo rozpuszczalne, które nie są wchłaniane przez ściany przewodu pokarmowego. Jest to podstawa do stosowania leków antymonu i bizmutu, na przykład zasadowego azotanu bizmutu. 51. Biologiczna rola pierwiastków p grupy VIA. Wykorzystanie ich związków w medycynie Zgodnie z zawartością w organizmie człowieka (62%), tlen należy do makroskładników. Jest niezbędny i jest jednym z najważniejszych elementów tworzących podstawę żywych systemów, czyli jest organogenem. Tlen jest częścią ogromnej liczby cząsteczek, od najprostszych po biopolimery. Rola tlenu w procesach życiowych jest ogromna, ponieważ utlenianie składników odżywczych (węglowodanów, białek, tłuszczów) tlenem służy jako źródło energii niezbędnej do funkcjonowania narządów i tkanek organizmów żywych. Większość reakcji redoks w organizmie zachodzi przy udziale tlenu i jego aktywnych form. Z obecnością tlenu związane są również funkcje fagocytarne (ochronne) organizmu, a zmniejszenie zawartości tlenu w organizmie obniża jego właściwości ochronne. W fagocytach (komórkach zdolnych do wychwytywania i trawienia ciał obcych) tlen O02 jest redukowany do ponadtlenku. W praktyce medycznej tlen stosuje się do inhalacji w stanach bolesnych, którym towarzyszy niedotlenienie (niedotlenienie), schorzeniach dróg oddechowych, układu krążenia, zatruciach tlenkiem węgla (II) CO, kwasem cyjanowodorowym HCN, a także schorzeniach z upośledzeniem funkcji układu oddechowego . Szeroko stosowane w praktyce klinicznej jest natlenianie hiperbaryczne - zastosowanie tlenu pod wysokim ciśnieniem. Alotropowa modyfikacja tlenu - ozonu O3 jako bardzo silnego środka utleniającego służy do dezynfekcji pomieszczeń, dezynfekcji powietrza i oczyszczania wody pitnej. Pod względem zawartości w organizmie człowieka (0,16%) siarka zaliczana jest do makroskładników. Podobnie jak tlen, jest niezbędny. Dzienne zapotrzebowanie osoby dorosłej na siarkę wynosi około 4-5 g. Siarka wchodzi w skład wielu biomolekuł - białek, aminokwasów (cystyna, cysteina, metionina itp.), hormonów (insulina), witamin (witamina B1). Dużo siarki zawarte jest w karotenie we włosach, kościach i tkance nerwowej. W organizmach żywych siarka, która jest częścią aminokwasów, jest utleniana. Produktami końcowymi tego procesu są głównie siarczany. Ponadto powstają tiosiarczany, siarka cementowa i kwasy politionowe. Zgodnie z zawartością w ciele (10-5-10-7%) selen jest mikroelementem. Niektórzy badacze uważają to za istotny element. Selen pochodzi z pożywienia - 55-110 mg rocznie. Selen koncentruje się głównie w wątrobie i nerkach. Stężenie selenu we krwi wynosi 0,001-0,004 mmol/l. Związek selenu z siarką w organizmach żywych jest niewątpliwy. W dużych dawkach selen gromadzi się przede wszystkim w paznokciach i włosach, które są oparte na aminokwasach zawierających siarkę. Znana jest również zdolność selenu do ochrony organizmu przed zatruciem rtęcią Hg i kadmem Cd. Selen promuje wiązanie tych toksycznych metali z innymi aktywnymi centrami, z tymi, na które nie ma wpływu ich toksyczne działanie. Ciekawostką jest związek między wysoką zawartością selenu w diecie a niską śmiertelnością z powodu raka. Selen jest toksyczny w dużych dawkach. Rozkład związków selenu u zwierząt prowadzi do uwolnienia wysoce toksycznego dimetyloselenu, który ma zapach czosnku. 52. Biologiczna rola pierwiastków p z grupy VIIA. Zastosowanie ich związków w medycynie (fluor i chlor) Pod względem zawartości w organizmie człowieka chlor (0,15%) należy do makroelementów, natomiast pozostałe pierwiastki tej grupy to mikroelementy (zawartość – 105 %). Halogeny w postaci różnych związków wchodzą w skład tkanek ludzi i zwierząt. Chlor i jod to pierwiastki niezastąpione, reszta to stałe składniki tkanek. Masa fluoru w organizmie człowieka wynosi około 7 mg (~105 %). Związki fluoru są skoncentrowane w tkance kostnej, paznokciach, zębach. W skład zębów wchodzi około 0,01% fluoru, a większość z niego opada na szkliwo, co wiąże się z obecnością w nim trudno rozpuszczalnego fluoroapatytu. Brak fluoru w organizmie prowadzi do próchnicy zębów. Zainteresowanie biologicznym działaniem fluoru wiąże się przede wszystkim z problemem chorób zębów, gdyż fluor chroni zęby przed próchnicą. Podstawą mineralną tkanek zęba (zębiny) jest hydroksyapatyt, chlorapatyt i fluoroapatyt. Bardzo często zniszczeniu ulega nie zewnętrzna powierzchnia zęba pokryta warstwą szkliwa, ale wewnętrzne części zębiny, odsłonięte w przypadku uszkodzenia szkliwa. Istnieją sugestie, że chociaż szkliwo jest lekko uszkodzone, wprowadzenie fluorku sodu sprzyja tworzeniu się fluoroapatytu, ułatwiając remineralizację rozpoczętych uszkodzeń. Fluorek sodu NaF jest stosowany w praktyce medycznej jako miejscowy środek zewnętrzny. Stosowanie NaF opiera się na tworzeniu fluoroapatytu. Jednocześnie dochodzi również do alkalizacji środowiska jamy ustnej, co przyczynia się do neutralizacji kwasów wytwarzanych przez bakterie. Szkodliwy jest nie tylko niedobór, ale i nadmiar fluoru. Gdy zawartość fluoru w wodzie pitnej jest wyższa niż maksymalna dopuszczalna norma (1,2 mg/l), szkliwo zębów staje się kruche, łatwo ulega zniszczeniu i pojawiają się inne objawy przewlekłego zatrucia fluorem - zwiększona łamliwość kości, deformacje kości i ogólne wyczerpanie organizmu . Choroba występująca w tym przypadku nazywa się fluorozą (fluorozą). Organizm ludzki zawiera około 100 g (2790 mmol) chloru. Chloridiony odgrywają ważną rolę biologiczną. Aktywują niektóre enzymy, tworzą sprzyjające środowisko dla działania enzymów protolitycznych soku żołądkowego, zapewniają przepływ jonów przez błony komórkowe oraz uczestniczą w utrzymaniu równowagi osmotycznej. Chloridion ma optymalny promień przenikania przez błonę komórkową. Tłumaczy to jego wspólny udział z jonami Na i K w tworzeniu określonego ciśnienia osmotycznego i regulacji metabolizmu wody z solą. Dzienne zapotrzebowanie na chlorek sodu wynosi 5-10 g. Jak już wspomniano, NaCl jest niezbędny do produkcji kwasu solnego w żołądku. Oprócz ważnej roli kwasu solnego w procesie trawienia, niszczy różne bakterie chorobotwórcze (cholera, dur brzuszny). Jeśli bakterie dostaną się do żołądka z dużą ilością wody, to dzięki rozcieńczeniu HCl nie działa on antybakteryjnie, a bakterie przeżywają. Prowadzi to do choroby w ciele. Dlatego podczas epidemii woda surowa jest szczególnie niebezpieczna. Przy niewystarczającej ilości kwasu solnego w żołądku wzrasta pH i zaburza się normalne trawienie, co poważnie wpływa na zdrowie człowieka. Przy zmniejszonej kwasowości soku żołądkowego w praktyce medycznej stosuje się rozcieńczony roztwór kwasu solnego. Przy zapaleniu żołądka (zapaleniu żołądka), wrzodzie trawiennym wzrasta wydzielanie soku żołądkowego, wzrasta jego kwasowość. 53. Biologiczna rola pierwiastków p z grupy VIIA. Zastosowanie ich związków w medycynie (brom, jod) Masa bromu w ludzkim ciele wynosi około 7 mg. Jest zlokalizowany głównie w gruczołach dokrewnych, przede wszystkim w przysadce mózgowej. Biologiczna rola związków bromu w normalnym funkcjonowaniu organizmu nie została jeszcze dostatecznie wyjaśniona. Związki bromu hamują pracę tarczycy i zwiększają aktywność kory nadnerczy. Kiedy bromidiony są wprowadzane do organizmu, centralny układ nerwowy jest najbardziej wrażliwy. Bromidiony równomiernie gromadzą się w różnych częściach mózgu i działają uspokajająco ze zwiększoną pobudliwością. Przyczyniają się do przywrócenia zaburzonej równowagi między procesami wzbudzania i hamowania. Bromidiony są łatwo wchłaniane w przewodzie pokarmowym. Toksyczność bromidionów jest niska. Ze względu na powolne wydalanie z organizmu (w ciągu 30-60 dni) mogą się kumulować (kumulować), co prowadzi do rozwoju przewlekłego zatrucia, zwanego bromizmem. W przypadku pojawienia się objawów przewlekłego zatrucia bromem preparaty bromowe należy natychmiast odstawić. Ponadto podaje się dużą ilość chlorku sodu (do 25 g dziennie) w celu zwiększenia szybkości uwalniania bromidionów (zasada Le Chateliera) i przepisuje się obfity napój. Ze względu na różne indywidualne wrażliwości dawkowanie preparatów bromu waha się od 0,05 do 2,0 g. Jod jest jednym z niezbędnych składników odżywczych, a jego związki odgrywają ważną rolę w procesach metabolicznych. Jod wpływa na syntezę niektórych białek, tłuszczów i hormonów. Organizm ludzki zawiera około 25 mg jodu. Z całkowitej ilości jodu w organizmie ponad połowa znajduje się w tarczycy. Prawie cały jod zawarty w tym gruczole występuje w stanie związanym (w postaci hormonów), a tylko około 1% ma postać jodidionu. Tarczyca jest zdolna do koncentracji I-25 razy w porównaniu z jego zawartością w osoczu. Tarczyca wydziela hormony tyroksynę i trójjodotyroninę. Niedoczynność tarczycy (niedoczynność tarczycy) może wiązać się ze zmniejszeniem jej zdolności do akumulacji jonów jodkowych, a także brakiem jodu w diecie (wole endemiczne). W przypadku wola endemicznego przepisuje się preparaty jodowe: (jodek potasu KI lub jodek sodu NaI) w dawkach odpowiadających dziennemu zapotrzebowaniu człowieka na jod (0,001 g jodku potasu). Na obszarach, gdzie występuje niedobór jodu, do soli kuchennej dodaje się NaI lub K, aby zapobiec endemicznemu wola! (1-2,5 g na 100 kg). Przy zwiększonej aktywności tarczycy (nadczynność tarczycy), z powodu nadmiernej syntezy hormonów tarczycy, obserwuje się nienormalnie zwiększone tempo procesów metabolicznych. Przy nieskuteczności tych leków w leczeniu nadczynności tarczycy stosuje się preparat radioaktywnego jodu 131 I, którego promieniowanie niszczy mieszki włosowe tarczycy, a tym samym zmniejsza nadmierną syntezę hormonów. Wszystkie pierwiastki z grupy VIIA są fizjologicznie aktywne, a chlor i jod są niezbędne do życiowej aktywności organizmu. Fluor uważany jest za pierwiastek niezbędny do normalnego funkcjonowania organizmów żywych. W ciele halogeny są wymienne, przy czym obserwuje się zarówno synergię, jak i antagonizm. 54. Aerozole Aerozole to układy zdyspergowane z gazowym medium dyspersyjnym. W zależności od stanu skupienia fazy rozproszonej wyróżnia się mgły - aerozole z ciekłą fazą rozproszoną; dymy, pyły – aerozole ze stałą fazą rozproszoną; smog - aerozole o mieszanej fazie rozproszonej. Rozmiary cząstek fazy zdyspergowanej aerozoli, zgodnie z klasyfikacją układów zdyspergowanych, wahają się od 107 do 109 m Podobnie jak inne układy dyspersyjne, aerozole uzyskuje się dwiema metodami: kondensacją i dyspersją. metoda kondensacji Fazę rozproszoną otrzymuje się z fazy gazowej w fizycznym procesie kondensacji cząsteczek do cząstek o wielkości koloidalnej. Metody dyspersji Cząstki o rozmiarach koloidalnych uzyskuje się poprzez mielenie większych kruszyw. Aerozole mają zdolność rozpraszania światła. Cząstki fazy rozproszonej aerozoli nie posiadają podwójnej warstwy elektrycznej, jednakże cząstki fazy rozproszonej bardzo często przenoszą ładunek elektryczny. Ładunek powstaje w wyniku tarcia lub w wyniku adsorpcji jonów gazu. Należy zauważyć, że bardzo często cząstki aerozolu (małe i duże) niosą ładunek o przeciwnym znaku. Rozdzielenie cząstek według wielkości w dużych objętościach aerozoli według wysokości może prowadzić do pojawienia się pola elektrycznego o dużym natężeniu. W ten sposób w chmurach następuje wyładowanie elektryczne - błyskawica. Aerozole są układami niestabilnymi kinetycznie i agregatowo, ponieważ na granicy faz nie ma podwójnej warstwy elektrycznej. Dlatego aerozole koagulują szybciej niż liozole. W medycynie aerozole stosuje się w terapii inhalacyjnej, w celu ochrony uszkodzonej skóry, dezynfekcji. Czasami powstawanie aerozoli jest wyjątkowo niepożądane. Aerozole niebezpieczne dla zdrowia ludzkiego powstają w odlewniach, ceramice oraz podczas wydobycia i przetwarzania różnych minerałów (rudy, węgla, azbestu itp.). Aerozole zawierające cząstki węgla powodują choroby płuc – antrakozę, tlenek krzemu (IV) – krzemicę, azbest – azbestozę. Choroby alergiczne wywoływane są przez aerozole utworzone przez pyłki roślin, pyły powstałe podczas przetwarzania bawełny, lnu, konopi itp. Zawiesiny bakterii, pleśni i wirusów – aerozole mikrobiologiczne lub bakteryjne – są jedną z dróg przenoszenia chorób zakaźnych: płuc gruźlica, grypa, ostre choroby układu oddechowego. Aerozole powstające podczas spalania paliw, których fazę rozproszoną stanowią sadza, żywice, popiół i rakotwórcze węglowodory, mają szkodliwy wpływ na organizm ludzki. Smog jest szczególnie niebezpieczny dla zdrowia. Dlatego też walka z pyłami i zanieczyszczeniami atmosfery staje się coraz ważniejsza. Oczyszczanie powietrza z aerozoli osiąga się poprzez wprowadzenie technologii bezodpadowych - wychwytywania cząstek fazy rozproszonej za pomocą filtrów, cyklonów (odpylaczy odśrodkowych) i pola elektrycznego wysokiego napięcia. 55. Emulsje Emulsje to układy mikroheterogeniczne, w których faza rozproszona i ośrodek dyspersyjny są cieczami nie mieszającymi się. Rozmiary cząstek fazy rozproszonej - kropelek cieczy - wahają się od 104 do 106 um. W zależności od stężenia fazy zdyspergowanej wyróżnia się emulsje: rozcieńczone, skoncentrowane i silnie stężone. W zależności od charakteru fazy zdyspergowanej i ośrodka dyspersyjnego wyróżnia się: 1) emulsje cieczy niepolarnej (DF) w cieczy polarnej (DS) - emulsje bezpośrednie, zwane emulsjami pierwszego rodzaju lub emulsjami typu „olej/woda” (O/W); 2) emulsje cieczy polarnej (DF) w niepolarnej (DS) - emulsje odwrócone, zwane emulsjami drugiego rodzaju lub emulsjami typu „woda/olej” (W/O). Tutaj DF i DS są odpowiednio fazą rozproszoną i ośrodkiem dyspersyjnym, „woda” to dowolna ciecz polarna, „olej” jest niepolarny. Rodzaj emulsji można ustawić: 1) pomiar przewodności elektrycznej; 2) mieszanie z nadmiarem cieczy polarnej lub niepolarnej; 3) barwienie barwnikami rozpuszczalnymi w wodzie lub w oleju; 4) przez zwilżenie i rozprowadzenie kropli emulsji na hydrofobowej lub hydrofilowej powierzchni. Emulsje, podobnie jak inne systemy dyspersyjne, mogą być: otrzymane metodami kondensacji i dyspersji. Emulsje w postaci grubych dyspersji są układami niestabilnymi kinetycznie i agregująco. Kiedy zderzają się kropelki rozproszonej fazy, łączą się (koalescencja). W wyniku koalescencji emulsja rozdziela się na dwie ciągłe fazy ciekłe. Aby zwiększyć stabilność emulsji, stosuje się stabilizatory - emulgatory. Są to środki powierzchniowo czynne, które w wyniku adsorpcji na granicy faz zmniejszają napięcie międzyfazowe i tworzą mocny mechanicznie film adsorpcyjny. Jeżeli emulgatorem jest jonowy środek powierzchniowo czynny, to przekazuje on kropelkom fazy rozproszonej ładunek elektryczny o tym samym znaku, w wyniku czego kropelki się odpychają. Rodzaj wytworzonej emulsji zależy od właściwości emulgatora. Medium dyspersyjne jest zawsze cieczą, która najlepiej rozpuszcza lub zwilża emulgator. Jako emulgatory stosuje się sole wyższych kwasów tłuszczowych, estry wyższych kwasów tłuszczowych i alkohole wielowodorotlenowe, aminy długołańcuchowe. Emulsje są powszechnie spotykane w przyrodzie. Emulsje to mleko, śmietana, śmietana, masło, żółtko jajka, mleczny sok roślinny, ropa naftowa. W medycynie szeroko stosowane są emulsje zawierające substancje lecznicze: pierwszy typ (O/W) do użytku wewnętrznego, drugi typ (W/O) do użytku zewnętrznego. Wiadomo, że tłuszcze roślinne i zwierzęce są lepiej przyswajalne przez organizm w postaci zemulgowanej (mleka). W tym przypadku pochodne kwasów cholowego i deoksycholowego działają jako emulgatory. Czasami istnieje potrzeba zniszczenia powstałej emulsji. Rozbicie emulsji nazywa się demulgacją. Demulgację przeprowadza się poprzez zwiększanie i obniżanie temperatury, działanie pola elektrycznego, wirowanie oraz dodatek elektrolitów i specjalnych substancji - demulgatorów. Demulgatory to środki powierzchniowo czynne o większej aktywności powierzchniowej niż emulgatory, ale nie posiadające zdolności tworzenia mechanicznie mocnej warstwy adsorpcyjnej. 56. Koloidalne środki powierzchniowo czynne Koloidalne środki powierzchniowo czynne to substancje, które z tym samym rozpuszczalnikiem, w zależności od warunków, tworzą roztwór rzeczywisty i koloidalny. Jak już wspomniano, cząsteczki środków powierzchniowo czynnych są amfifilowe. Składają się z grup niepolarnych i polarnych. Rodniki niepolarne, na przykład łańcuchy węglowodorowe, nie mają powinowactwa do polarnego rozpuszczalnika - wody, w przypadku grup polarnych jest ono dość wysokie. Pomiędzy grupami niepolarnymi zachodzi oddziaływanie hydrofobowe (van der Waalsa). Z łańcuszkiem o długości około 1022 atomy węgla w wyniku oddziaływań hydrofobowych rodników węglowodorowych i silnego oddziaływania grup polarnych z wodą wiążą cząsteczki środka powierzchniowo czynnego i tworzą się micele. Minimalne stężenie koloidalnego środka powierzchniowo czynnego, od którego w jego roztworze tworzą się micele, nazywane jest krytycznym stężeniem micelarnym (CMC). Kształt powstałych miceli zależy od stężenia roztworu. Przy niskich stężeniach koloidalnego środka powierzchniowo czynnego tworzą się sferyczne micele. Wzrost stężenia roztworu koloidalnego środka powierzchniowo czynnego prowadzi najpierw do wzrostu ich liczby, a następnie do zmiany kształtu. W wyższych stężeniach zamiast miceli kulistych powstają micele cylindryczne i lamelarne. Wartość CMC zależy od różnych czynników: charakteru koloidalnego środka powierzchniowo czynnego, temperatury oraz obecności zanieczyszczeń obcych substancji, zwłaszcza elektrolitów. CMC można określić na podstawie właściwości roztworu w zależności od liczby i wielkości cząstek kinetycznie aktywnych, w szczególności na podstawie zmian ciśnienia osmotycznego, napięcia powierzchniowego, przewodności elektrycznej i właściwości optycznych. Ponieważ podczas przejścia „roztwór rzeczywisty – roztwór koloidalny” zmienia się wielkość cząstek aktywnych kinetycznie (jonów, cząsteczek, miceli) oraz ich liczba, na wykresie „właściwość – stężenie” pojawia się punkt przegięcia odpowiadający CMC. Jedną z najważniejszych właściwości roztworów koloidalnych środków powierzchniowo czynnych, dzięki której znajdują szerokie zastosowanie w różnych sektorach gospodarki narodowej oraz w medycynie, jest solubilizacja. Mechanizm solubilizacji polega na rozpuszczeniu substancji niepolarnych w hydrofobowym rdzeniu miceli. Zjawisko solubilizacji ma szerokie zastosowanie w różnych sektorach gospodarki narodowej: w przemyśle spożywczym, w przemyśle farmaceutycznym (do otrzymywania płynnych form substancji leczniczych). W układzie woda-fosfolipidy potrząsając i mieszając tworzą się kuliste micele - liposomy. Cząsteczki fosfolipidów tworzą w liposomach dwuwarstwową membranę, w której grupy polarne są zwrócone w stronę wody, a grupy niepolarne naprzeciw siebie. Liposomy można uznać za model błon biologicznych. Za ich pomocą można badać przepuszczalność membran i wpływ na nią różnych czynników dla różnych związków. Liposomy są szeroko stosowane do celowanego dostarczania leków do określonych narządów lub obszarów dotkniętych chorobą. Liposomy mogą transportować leki do komórek. Błony liposomalne są wykorzystywane w badaniach immunologicznych do badania interakcji między przeciwciałami a antygenami. Autorzy: Drozdova M.V., Drozdov
▪ Historia myśli ekonomicznej. Kurs wykładowy ▪ Pedagogika społeczna. Kołyska ▪ Pieniądze, kredyty, banki. Kołyska
Nowy sposób kontrolowania i manipulowania sygnałami optycznymi
05.05.2024 Klawiatura Primium Seneca
05.05.2024 Otwarto najwyższe obserwatorium astronomiczne na świecie
04.05.2024
▪ sekcja witryny Podstawy pierwszej pomocy (OPMP). Wybór artykułu ▪ artykuł Dziękuję, nie spodziewałem się ... Popularne wyrażenie ▪ artykuł Kto jest właścicielem zarówno Nagrody Nobla, jak i Ig Nobla? Szczegółowa odpowiedź ▪ artykuł Różowy oset. Legendy, uprawa, metody aplikacji ▪ artykuł japoński lakier. Proste przepisy i porady ▪ artykuł Ładowarka z timerem. Encyklopedia elektroniki radiowej i elektrotechniki
Strona główna | biblioteka | Artykuły | Mapa stony | Recenzje witryn
www.diagram.com.ua |


 Zobacz inne artykuły Sekcja
Zobacz inne artykuły Sekcja 